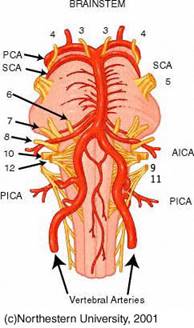
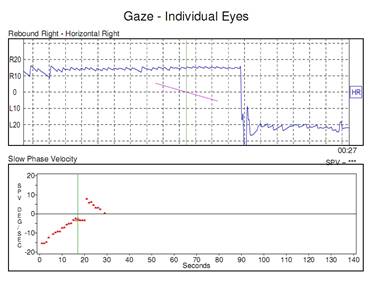
MALATTIA DEI PICCOLI VASI: PICA AICA
SINDROME DELL'ARTERIA LABIRINTICA SCA
Cause di ictus del tronco
encefalico - Le basi
l’ictus (Stroke) può
essere ischemico (mancanza di flusso sanguigno) o emorragico
(fuoriuscita di sangue nel cervello).
FATTORI DI RISCHIO PER TIA E L’ICTUS
To some extent, one can
predict risk of stroke.
In una certa misura, si può predire il rischio di ictus. Risk factors that are well known include: I fattori
di rischio che sono ben noti includono:
High Blood Pressure
(2)
Alta pressione sanguigna (2)
Collagen Vascular
disease
Malattie vascolari del collagene
Heart problem such as
atrial fibrillation (1.5) or old infarction (2.7) Problema
di cuore come la fibrillazione atriale (1.5) o vecchio infarto (2,7)
**Numbers in () are derived from
Whisnant et al, 1996** I
numeri in () sono derivati da Whisnant et al, 1996 Rischio di
pressione sanguigna elevata è ripida e chiaro.
Rischio dovuto
alla pressione sanguigna. Risk from elevated blood
pressure is steep and clear.Ad esempio, nello studio UK TIA, il rischio di recidiva di
ictus è aumentato del 28% per ogni aumento di 10 mm Hg della pressione
sistolica tra 130 e 160 (Farrell et al, 1991).
Although
LDL-cholesteral, HDL cholesteral seems to reduce risk of stroke (Sacco et al,
2001).Anche se
LDL-colesterolo, HDL colesterolo sembra ridurre il rischio di ictus (Sacco et
al, 2001).If your HDL cholesterol is > 35, then subtract one risk factor (a
negative risk factor).Se il
colesterolo HDL è> 35, bisogna sottrarre un fattore di rischio (un fattore
di rischio negativo).Mitral valve prolapse is not a significant risk factor, overall (0.8 risk). Il prolasso della valvola
mitrale non è un fattore di rischio significativo, nel complesso (rischio x 0,8).TIA is a
very strong risk factor for stroke (5.6 x risk).TIA è un fattore di rischio molto forte
per l'ictus (rischio x 5.6).In general, relative risk for most of the
above factors decreases with age (Whisnant et al, 1996), lending support for a
unaggressive approach to risk factors in individuals of advanced age.In generale, il rischio
relativo per la maggior parte dei fattori di cui sopra diminuisce con l'età
(Whisnant et al, 1996), sostenendo un approccio non aggressivo per i fattori di
rischio in individui di età avanzata.
Risk from
cholesterol can be further stratified into three groups, based on LDL (total
cholesterol - HDL)-(triglycerides).Rischio da colesterolo può essere ulteriormente
stratificata in tre gruppi, sulla base LDL (colesterolo totale - HDL) -
(trigliceridi).
LDLLDL
Risk FactorsFattori di rischio
Risk Level for vascular diseaseLivello di
rischio per malattia vascolare
< 130
<130
None
Nessuno
Low
Basso
130-159
130-159
Less than 2 Meno di 2
Moderate
Moderato
>130
> 130
more than 2 più di 2
High
Alto
Controllable
risk factors include being overweight, having high (> 140/90) or low blood
pressure, heart disease, diabetes and smoking.Fattori di rischio controllabili comprendono
il sovrappeso, con elevata (> 140/90) o bassa pressione sanguigna, malattie
cardiache, diabete e fumo.Atrial fibrillation is a particularly
important risk factor -- stroke occurs in 4.5% of untreated patients with
atrial fibrillation per year.La fibrillazione atriale è un fattore di rischio
particolarmente importante – l’ictus si verifica nel 4,5% dei pazienti trattati
con fibrillazione atriale all'anno.While uncommon, chiropractic neck
manipulations can cause compression or tears of the vertebral arteries (Vibert
et al, 1993; Smith et al, 2003), and for this reason, maneuvers involving neck
"cracking" should be specifically avoided in individuals with
vertigo.Mentre ,
le manipolazioni chiropratiche del collo possono causare compressione o rotture
delle arterie vertebrali (Vibert et al, 1993; Smith et al, 2003) e per questo
motivo, le manovre "cracking" che coinvolgono il collo devono essere
specificamente evitati nei soggetti con vertigine.Whiplash
injuries can also damage the vertebral arteries as the arteries traverse the
vertebrae of the neck.
Il colpo
di frusta può anche danneggiare le arterie vertebrali che passano attraversano
le vertebre del collo.
L’ictus
ischemico è causato da ostruzione dei vasi sanguigni. L’ostruzione
può provenire da una sorgente distante - come ad esempio un coagulo dal cuore –
che poi viene chiamato "embolo". I blocchi possono derivare da
coagulazione dall'interno dei vasi - che poi sono chiamati "trombi".
Gli ictus trombotici sono più spesso attribuiti a accumulo di colesterolo
all'interno delle pareti del vaso sanguigno, che producono turbolenza e ispessimento
irregolare della parete, con successiva e conseguente formazione di coaguli.
Un'altra causa comune di ictus trombotico è una caduta di pressione sanguigna
- come potrebbe essere ad esempio causata da un arresto cardiaco di alcuni
minuti. Al di fuori di queste due categorie generali (emboli e trombi)ci possono
essere solo fonti occasionali di ictus ischemico Ad esempio, causati da
ostruzione meccanica dei vasi sanguigni – come potrebbe accadere durante una manipolazione
chiropratica del collo ad alta velocità o qualche altro evento che
causa un movimento collo molto forte. Si sono riscontrati (ad esempio), ictus
in pazienti subito dopo che gli stessi erano stati sulle montagne russe.
Ictus
emorragici - fuoriuscita di sangue all'interno del cervello - sono più
comunemente causate da pressione sanguigna troppo alta. Altre possibili
cause comprendono irregolarità nella coagulazione del sangue, danni alle pareti
dei vasi sanguigni da vari processi (compreso l'ictus ischemico). Nel
tronco encefalico, il circuito è stretto e gli ictus emorragici sono spesso
devastanti.
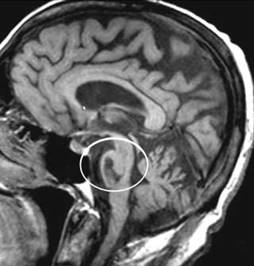
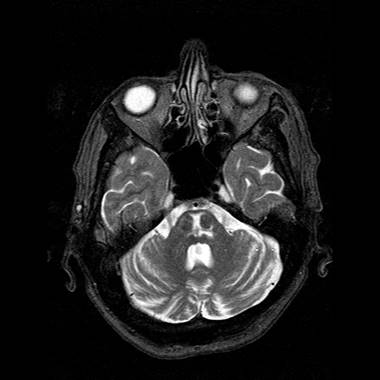
La sindrome PICA è
conosciuto anche come "sindrome laterale midollare", o "sindrome
di Wallenberg", dopo la descrizione di Wallenberg nel 1895. Questo è il
colpo del tronco cerebrale più comune. Vedere questo
link per maggiori dettagli.
La sindrome AICA è di solito
accompagnata da vertigini e sordità omolaterale unilaterale dalla labirintica
ischemia dell'arteria. Si tratta di un ictus del tronco cerebrale comune. Vedere questo
link per maggiori dettagli.
Sindrome dell'arteria
labirintica
L'arteria
uditiva labirintico o interno di solito prende la sua origine da AICA, ma può
anche prendere origine dal PICA o l'arteria basilare. Fornisce l'orecchio
interno. Nel canale uditivo interno o IAC fornisce ganglio di
Scarpa. Dopo l'uscita si divide in arteria cocleare comune e anteriore
arteria vestibolare. L'arteria cocleare comune ulteriore divide in arteria
cocleare e l'arteria vestibolococleare, quest'ultima formando posteriore
arteria vestibolare e il ramo vestibolare. La principale arteria cocleare
fornisce il apicale 3/4 della coclea, e il ramo cocleare, le basali 1/4 (alte
frequenze). L'arteria vestibolare posteriore fornisce il sacculo inferiore
e l'ampolla del SCC posteriori. L'arteria vestibolare anteriore è una arteria
minore che fornisce utricolo, sacculo superiore, ed ampolla anteriore e nei canali
semicircolari laterali (Kim et al, 1999). L'arteria labirintica è un arteria
sottile terminale e come tale può essere relativamente più vulnerabile di altre
circolazioni.
La
diagnosi può essere difficile perché il cervello può mostrare nessuna lesione. Questo
è un piccolo vaso sanguigno e studi di imaging come TAC-angiografica o RMA
perdere. Il contrasto è assolutamente necessario (durante MRA).
I
sintomi principali sono atassie cerebellari omolaterali (peduncoli cerebellari
medi e / o superiori), nausea e vomito, impastata (pseudobulbare) discorso, la
perdita del dolore e della temperatura sul lato opposto del corpo. Sordità
parziale, tremore degli arti superiori, una sindrome di Horner omolaterale
e mioclono
palatale sono stati segnalati. Clinicamente, questo colpo può
essere impossibile da distinguere da una AICA parziale o ictus territorio
PICA. E 'molto più raro di uno dei due. Pulsione oculare lontano dal
lato della lesione è stata riportata nella sindrome SCA. Diagnosi della
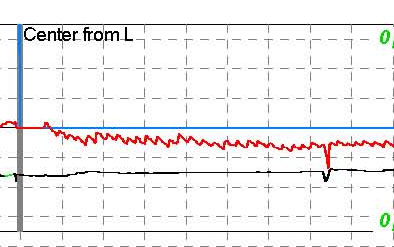
corsa avviene tramite risonanza magnetica.
Nella
sindrome di Weber, vi è un danno per il mesencefalo, compresi i fascicoli del III°
nervo all'interno del mesencefalo e anche lì vi è un danno al fascio piramidale
prima della decussatione (area del peduncolo cerebrale). Ciò si traduce in
una paralisi del III° nervo omolaterale combinata con una emiplegia
controlaterale. Questa sindrome può derivare da danni alla SCA.
Sindrome di Weber
Infarto
del mesencefalo laterale risultante da un aneurisma dell'arteria cerebellare
superiore. La lesione è qui solo posteriormente al peduncolo cerebrale.
Emorragia Pontine.
Questo è un evento
catastrofico, tipicamente un spurgo ipertensiva. Si presenta con di coma,
quadriplegia, piccole pupille reattive e assenti movimenti oculari
orizzontali. Nella maggior parte dei pazienti tetraplegici un ematoma nel
mezzo del ponte è centrata all'incrocio tra tegmento e la base
Pontis. Bobbing oculare è una caratteristica costante. Emorragie
presenti con 1 1/2 sindrome, piccole pupille reattive, arti atassia cerebellare
di tipo, e la perdita di hemisensory controlaterale (Caplan e Goodwin, 1982)
tegmentale laterale. Quelli che sopravvivono possono sviluppare mioclono
oculopalatal . La diagnosi può essere effettuato tramite MRI
(migliore) o TC, o una combinazione di entrambi.
Medial infarto midollare
(sindrome di Dejerine).
0,5% di tutte infarti
cerebrali. Emiparesi controlaterale risparmiando il volto, perdita
hemisensory del tipo colonna posteriore (controlaterale). Debolezza della
lingua è omolaterale al infarto. Patologia può essere in arteria
vertebrale o di un arto mesiale dell'arteria vertebrale dopo PICA. nistagmo Upbeat può
verificarsi. Malattia dei piccoli vasi (diabete, ipertensione,
ipercolesterolemia) è la causa usuale.
Risonanza magnetica di persona con
Mielinolisi pontine centrale, vista assiale. Notare la "I"
zona a forma al centro del ponte.
Lesioni iperintense Pontine.
E 'comune incontrare zone di
maggiore segnale T2 RMN nel ponte nelle persone anziane con
instabilità. Questi pazienti mostrano spesso i sintomi di squilibrio,
difficili con il discorso e la deglutizione. (Kwa et al,
1998). Secondo l'esperienza dell'autore, questi pazienti spesso mostrano nistagmo da rimbalzo ,
che è una variante di nistagmo evocato
sguardo- .
Una rara fonte di lesioni
iperintense pontine è mielinolisi
pontine centrale (vedi sopra). Questo è causato da rapide fluttuazioni
nello stato elettrolita, generalmente nell'ambito di un
ricovero. L'individuo indicato sopra ha avuto un trapianto di fegato
fatto. Dopo il trapianto di fegato, è stato bene per un paio di giorni, ma
poi è diventato a poco a poco in coma. La sua risonanza magnetica in quel
momento ha mostrato sopra l'immagine. Esame nove mesi più tardi rivelato
un individuo deambulatorio con alcuni segni cerebellari miti. Circa il 2%
delle persone con trapianto di fegato sviluppare mielinolisi pontina centrale.
Gli individui con infarti
linea mediana pontine hanno di solito normale test ABR (faught
e Oh, 1985).
References:
Adams,
Victor and Ropper. Principles of Neurology 6th edn, McGraw Hill, 1997
Bradshaw P,
McQuaid P. The syndrome of vertebro-basilar insufficiency. Q.J.Med
1963:32:279-296
Caplan LR.
Vertebrobasilar disease. In Barnet HJM (and others, Eds), Stroke:
Pathophysiology, Diagnosis and Management. New York:
Chrchill-Livingstone, pp 549-619, 1986
Faught E,
Oh SJ. Brainstem auditory responses in brainstem infarction. Stroke
1985. 16:701-705
Fisher CM.
Vertigo in cerebrovascular disease. Arch Otolaryngol 1967;85:529-534
Grad A,
Baloh RW. Vertigo of vascular origin: clinical and electronystagmographic
features in 84 cases. Arch Neurol 1989;46;281-284
Kim JS and
others. Internal auditory artery infarction. Neurology
1999:42:40-44
Kubik CS,
Adams RD. Occlusion of the basilar artery: a clinical and pathological
study. Brain
1946: 69:73-121
Kwa VIH,
Zaal LH, Verbeeten B, Stam J. Disequilibrium in patients with
atherosclerosis. Relevance
of pontine ischemic infarction. Neurology 1998;51:570-573.
Wallenberg
A. Akute bulbar affektion. Arch Psychiatr Nervenheilkd
1895:27:504-540
William D,
Wilson TG. The diagnosis of the major and minor syndromes of basilar
insufficiency. Brain
1962;85:741-774
Toyoda et
al. Medial Medullary Infarction: Analysis of eleven patients. Neurology
1996:47:1141-1147
PICA
(sindrome dell'arteria posteriore cerebellare). o Infarto bulbare laterale (s.
di Wallemberg)
La sindrome PICA è conosciuto anche come "sindrome laterale
midollare", o "sindrome di Wallenberg"o
“sindrome dell’arteria cerebellare postero-inferiore”, dopo la descrizione di
Wallenberg nel 1895.Dipende da un’occlusione o dell’arteria cerebellare
posteroinferiore (PICA) o del tratto terminale di un’arteria vertebrale da
dove questo ramo origina. La lesione ischemica interessa il segmento
dorso-laterale del bulbo (Fig. 4A-B). Questo è l’ictus più comune del
tronco cerebrale.
Fig.
4-
A)
sede della lesione infartuale defla 5. ischemica lat. del bulbo (s. di
Wallemberg) (da Baloh R.W., 1984); B) sez. trasversa del bulbo che illustra la
distribuzione dell’a. cerebellare post. inf. (da Silver F.L. in Sharpe J. e
Barber H. Ed., 1993);C) sez. trasversa della zona dorso laterale bulbo-pontina
irrorata dall’a. cerebellare ant.-inf. (da Silver F. L. in Sharpe J. e Barber
H. Ed., 1993). da Babighia Otoneurologia Piccin2008
SINTOMATOLOGIA
La
sintomatologia compare all’improvviso ed è caratterizzata da vertigine assai
intensa, da perdita di equilibrio con latero-pulsione sul lato della lesione
(emiatassia omolaterale), da disartria, ptosi e miosi, da cefalea occipitale e
da sintomi neurovegetativi ipervagotonici..All’inizio, l’episodio
vertiginoso può venir confuso con quello di una lesione acuta del labirinto o
delle strutture nervose che appartengono all’arco primario vestibolare. Il
nistagmo può essere unico, di tipo orizzontale, in genere diretto verso il lato
opposto alla sede della lesione oppure multiplo. In tal caso, al nistagmo
“paralitico” si associa un nistagmo da “direzione dello sguardo”,
prevalentemente orizzontale- rotatorio o rotatorio, diretto verso il lato del
focolaio ischemico. Compaiono ampi movimenti sacca- dici (ipermetrici), sia
volontari che involontari, diretti verso il lato della lesione mentre quelli
diretti verso il lato opposto sono molto piccoli. Anche i movimenti oculari
lenti di conduzione (pursuit) appaiono asimmetrici. Oltre ai segni di
sofferenza cerebellare: asinergia, atassia, adiadococinesi (per
l’interessamento del distretto postero-inferiore dell’emisfero cerebellare e
del verme), si osservano disfonia e disfagia per paralisi omolaterale del X e
del 1X, ipoestesia dell’emifaccia con riduzione o scomparsa del riflesso
corneale per interessamento del nucleo e della radice discendente del V,
paralisi omolaterale del VII. Frequente è la comparsa della sindrome di Claude
Bernard-Horner (miosi, enoftalmo, ptosi palpebrale) omolaterale alla sede della
lesione per interessamento delle vie simpatiche oculomotorie che corrono in
questo distretto bulbare. Nel lato opposto alla lesione, si osserva una
emianestesia dissociata termo-dolorifica.
Il
nistagmo provocato da stimolo calorico presenta, oltre alle modificazioni
qualitative delle scosse (prevalenza della fase lenta, aritmia, variazioni
della forma), un’alterazione dei valori dei parametri quantitativi che varia in
rapporto all’estensione della lesione vascolare e, soprattutto, al grado di
interessamento dell’area nucleare vestibolare o dei distretti nervosi
iuxtanucleari. Nella sindrome di Wallemberg, come in altre lesioni che
interessano il segmento del bulbo posteriore all’oliva, non è possibile mettere
in evidenza, nel corso della reazione vestibolare, la deviazione tonica degli
arti superiori (prova di Bàràny) per l’interessamento delle vie di connessione
vestibolo-spinali. Nel corso della reazione vestibolo-oculomotoria provocata
dallo stimolo calorico, non si osserva la comparsa del “perverted nystagmus”.
Le
scosse del NOC presentano alcune alterazioni morfologiche legate all’
interferenza del nistagmo spontaneo o del nistagmo da “direzione dello sguardo”
e alla presenza di anomalie dei saccadici soprattutto di quelli diretti verso
il lato della lesione.
La
maggior parte dei pazienti dopo questo ictus recupera molto bene e spesso
riprendono le loro precedenti attività (Nelles et al, 1998). I pazienti
spesso hanno la sindrome di Horner (ptosi monolaterale, miosi e anidrosi
facciale). Ci possono essere anche saccadici dismetrici (overshoot),
pulsione saccadici (tirando dell'occhio durante le saccadi verticali verso il
lato di lesione).La prognosi è generalmente molto buono con recupero pieno o
quasi completo atteso entro i 6 mesi. La diagnosi si fa generalmente con
la risonanza magnetica. CT-angiografia con ricostruzione in 3D ha ottenuto
abbastanza buono in questi ultimi anni per essere utile troppo.
L’esame ABR è spesso anormale nelle persone con una sindrome di
Horner centrale (Faught e Oh, 1985), ma siccome la lesione nella sindrome di
Wallenberg è abitualmente inferiore alle vie uditive, la sindrome di Horner
prodotta dalla Wallenberg generalmente non è associata ad ABR anormali.
La PICA deriva normalmente dall'arteria vertebrale , oppure da un
ramo distinto dell'arteria basilare. A causa dell'origine molto più
comune dalla arteria vertebrale, la maggior parte delle sindrome (ictus)
"PICA" in realtà sono dovuti ad occlusione dell'arteria vertebrale
(Kim 2003) l’embolia cardiaca provoca solo il 5% di questi ictus , mentre la
dissezione provoca il 15% (Kim, 2003).
PICA è il sito più comune di occlusione dalla moltiplicazione dei trombi
o embolie causata da lesioni alla terza sezione dell'arteria vertebrale, e la
sindrome di Wallenberg è la patologia più comune causata da manipolazione
chiropratica (Caplan, 1986).
References
Caplan LR.
Vertebrobasilar disease. In Barnet HJM (and others, Eds), Stroke:
Pathophysiology, Diagnosis and Management. New York:
Chrchill-Livingstone, pp 549-619, 1986
Faught E,
Oh SJ. Brainstem auditory responses in brainstem infarction. Stroke
1985. 16:701-705
Kim JS
(2003). "Pure lateral medullary infarction: clinical-radiological
correlation of 130 acute, consecutive patients." Brain
126(Pt 8): 1864-72.
Nelles G,
and others. Recovery following lateral medullary infarction. Neurology
1998:50:1418-1422
AICA (arteria
cerebellare antero inferiore)sindrome.
L'arteria cerebellare anteriore inferiore(AICA)
è un'arteria nel cervello che fornisce parte del cervelletto .
Esso deriva dalla arteria basilare a livello della giunzione tra
il midollo allungato ed i ponte nel tronco cerebrale . Si passa all'indietro da
distribuire alla parte anteriore della superficie inferiore del cervelletto,
anastomosi con il posteriore inferiore cerebellare ramo dell'arteria vertebrale
. Fornisce il quartiere inferiore anteriore del cervelletto.
Dà anche origine, nella maggior parte dei casi dell'arteria
labirintica; Tuttavia, in altri casi l'arteria labirintica può emergere come un
ramo della basilare. La sindrome AICA solito si presenta con vertigini e
sordità omolaterale unilaterale dalla labirintica ischemia dell'arteria. I
grandi ictus sono accompagnati da paralisi facciale omolaterale e atassia. E '
per frequenza il secondo più comune ictus del tronco cerebrale, circa il 10% degli
ictus colpiscono la PICA.
L'entità di questo tratto è
estremamente variabile. Sintomi simili a quelli della malattia
di Meniere (udito fluttuante, acufeni, vertigini) possono essere
causati anche da TIA in tale distribuzione (Lee e Cho, 2003). La bilateralità
delle fluttuazione uditive suggerisce una causa vascolare, ma la maggior parte
degli ictus da AICA si presentono con sintomi uditivi unilaterali. L'AICA
ha anche una origine molto variabile e può prendere origine dalla linea caudale
mediana del ponte. Gli ictus del territorio dell’AICA possono
presentare anche solo vertigini . La diagnosi si ottiene generalmente
tramite risonanza magnetica.
References
Lee H, Cho
YW. Auditory disturbance as a prodrome of anterior inferior cerebellar
artery infarction. J.
Neurol Neurosurg Psych 2003:74:1644-1648
Tab. I - Sintomi e strutture nervose interessate nelle
lesioni infartuali nel territorio
irrorato dalla P1CA e da AICA
Sintomi
Infarto
nel
territorio
della PICA
Infarto
nel
territorio
della AICA
Vertigine
Nn. vest., distretto cereb.
post-inf.
Distretto
dorsolat. b-pontino, flocculo, labirinto
Parole
chiave : Vasculite, Sistema nervoso centrale,
Cervello, Angioite cerebrale, Sindrome di vasocostrizione cerebrale reversibile
Riassunto
La diagnosi di vasculite del sistema nervoso centrale (SNC) è
difficile e spesso posta per eccesso, in assenza di conferma istologica. Le
manifestazioni cliniche delle vasculiti del SNC sono proteiformi e totalmente
aspecifiche. Le indagini radiologiche cerebrovascolari, in particolare
l'angio-risonanza magnetica e/o l'arteriografia cerebrale convenzionale,
possono mostrare delle immagini suggestive tipo stenosi multifocali e/o
aneurismi multipli, ma anche in questo caso non specifiche. Diverse altre
diagnosi devono essere scartate, in particolare una sindrome di vasocostrizione
cerebrale reversibile e/o delle lesioni di aterosclerosi intracranica, che
possono fornire degli aspetti molto simili alla diagnostica per immagini. Sole
una biopsia leptomeningea, raramente realizzata in pratica, può confermare la
diagnosi di vasculite, ma non permette sempre di determinare il suo carattere
primitivo o secondario (in particolare a una malattia sistemica o a
un'infezione). Le vasculiti cerebrali primitive restano eccezionali, con cause
e meccanismi totalmente sconosciuti, oltre 50anni dopo le loro prime
descrizioni. L'esclusione di tutte le diagnosi differenziali e di tutte le
cause secondarie di vasculite del SNC è indispensabile, ma può essere
complessa, tanto più che l'instaurazione di una terapia adeguata non deve
essere troppo ritardata. Anche se la prognosi di queste vasculiti cerebrali
primitive è migliore oggi rispetto a 50anni fa, grazie all'utilizzo di
corticosteroidi e immunosoppressori, le sequele cognitive sono frequenti. Sono
attualmente necessari degli studi multicentrici prospettici per definire lo
schema terapeutico ottimale, adattato in funzione delle caratteristiche di ogni
paziente.
Il testo completo di questo articolo è disponibile in PDF.
In queste patologie (esempi comuni:
arteriolosclerosi e vasculopatia diabetica) l’ipoafflusso ematico è
generalizzato a tutto l’encefalo ed i segni possono variare secondo i distretti
più colpiti. Le forme di più frequente osservazione riguardano pazienti
anziani, nei quali la sintomatologia assume i caratteri di disorientamento
spaziale e di disordine motorio piuttosto che di vertigine. Spesso
l’associazione di disturbi della visione e della propriocezione periferica
aggravano il disagio soggettivo. In questi casi l’indagine vestibolare può
essere utile per stabilire una diagnosi funzionale in vista di programmi
riabilitativi motori. Le prove funzionali possono svelare una iporeflessia
vestibolare bilaterale scarsamente compensata per la coesistenza di difetti
visivi e di propriocezione. Il Ny indotto da stimolazioni labirintiche può
rivelarsi a micro scrittura” (alta frequenza ed ampiezza ridotta), scarsamente
inibito o addirittura potenziato dalla fissazione visiva, segni questi
considerati indicativi di disinibizione da sofferenza corticale diffusa. Spesso
in questi pazienti una RMN mostra uno stato più o meno marcato di atrofia
corticale.
E dubbio se una lesione emisferica possa
indurre una vertigine. Si ammette questa possibilità sulla base di classiche
esperienze che hanno dimostrato che in seguito alla stimolazione elettrica
della corteccia del lobo temporale e del lobo parietale vicino alla scissura
silviana si inducono vertigini intense, Nelle lesioni emisferiche la vertigine
è spesso associata ad afasia. emiparesi, emisindrome sensitiva. emianopsia.
Le vasculiti del Sistema Nervoso (CNS) Centrale rappresentano un gruppo eterogeneo di malattie
infiammatorie che interessano le pareti dei vasi sanguigni nel cervello,
midollo spinale, e le meningi.
Si prega di fare riferimento all'articolo su vasculite per una
discussione generale di tale entità.
Lo scopo di questo articolo è quello di discutere la angioite primaria del sistema nervoso centrale (PACNS) dal momento che le altre vasculiti sono già discussi in
articoli specifici.
Terminologia
Vasculiti del SNC sono classificati come 1-2:
·primario: confinata al SNC, senza coinvolgimento di altri
sistemi - denominato come PACNS
·secondaria: si verifica nel contesto di un processo
infiammatorio o sistemica infettive
Si prega di notare che questa classificazione è diverso da
quello usato quando si parla di vasculiti sistemiche.
Epidemiologia
PACNS rimane una malattia rara: un tasso di incidenza stimato
medio annuo di 2,4 casi per milione. Essa colpisce i pazienti di tutte le
età, ma picchi a circa 50 anni di età, con i maschi colpiti più comunemente
rispetto alle femmine 1.
Cause secondarie di CNS vasculite superano di gran lunga in
numero PACNS 2. Si prega di fare riferimento a ciascuna
vasculite specifiche per ulteriori dettagli.
La presentazione clinica
Caratteristiche cliniche della PACNS sono aspecifici. La
diagnosi viene fatta in base a criteri di Calabrese 4, tra
cui:
·presenza di un deficit neurologico o psichiatrico altrimenti
inspiegabile acquisiti
·presenza di entrambi i classici caratteristiche angiografiche o
istopatologiche del angioite all'interno del sistema nervoso centrale (biopsia
rimane lo standard di riferimento per la diagnosi 3)
·nessuna evidenza di vasculite sistemica o qualsiasi disturbo che
potrebbero causare o imitare le caratteristiche angiografiche o patologici
della malattia
Quando parte di un disturbo sistemico, la diagnosi può essere
più facile, a meno che i sintomi cerebrali sono i primi a manifestare. Si
prega di fare riferimento a una vasculite specifica per ulteriori dettagli
sulla manifestazione clinica.
Patologia
Per quasi tutte le forme di vasculite, compreso PACNS, il
fattore scatenante è sconosciuto3.
I risultati di imaging per PACNS sono generalmente variabili e
non specifici, con infarti ischemici le
lesioni più comuni, che si verificano nel 53% dei casi 5.
CT
Può mostrare aree di hypoattenuation.
MRI
Più specifico per mostrare più infarti: solitamente bilaterale,
che interessano vari territori vascolari di dimensioni variabili, e in vari
stadi di guarigione.
T2 e FLAIR ad alta intensità lesioni della sostanza bianca sono
molto comuni in PACNS, ma completamente non specifica.
Spettacoli focale o multifocale segmentale restringimento dei
vasi sanguigni sia di piccole e medie dimensioni, sono presenti anche le occlusioni. Gli
stessi risultati potrebbero essere dimostrati in entrambi CTA e MRA.
Trattamento e la prognosi
PACNS è gestito con steroidi ad alto dosaggio e agenti
citotossici 3.
Storia ed etimologia
La PACNS è stata inizialmente presentata nel 1959 da Humberto Cravioto e Irwin Feigin6.
Ricordate che, nonostante di essere composta da reperti non
specifici, la RM è quasi il 100% sensibile per PACNS e un esame normale esclude
praticamente questa diagnosi 1.
·4.
Calabrese LH, Mallek JA. Primary angiitis of the central nervous system. Report
of 8 new cases, review of the literature, and proposal for diagnostic criteria.
Medicine
(Baltimore). 1988;67 (1): 20-39. Pubmed citation
·5.
Salvarani C, Brown RD, Calamia KT et-al. Primary central
nervous system vasculitis: analysis of 101 patients. Ann. Neurol.
2007;62 (5): 442-51.doi:10.1002/ana.21226 - Pubmed citation
·6.
Cravioto H, Feigin I. Noninfectious granulomatous angiitis with a predilection
for the nervous system. Neurology.
1998;9: 599-609. Pubmed citation
The central nervous system (CNS) may be involved by a variety of
inflammatory diseases of blood vessels. Il sistema nervoso
centrale (CNS) può essere coinvolto da una varietà di malattie infiammatorie
dei vasi sanguigni. These include primary angiitis
of the central nervous system (PACNS), a rare disorder specifically targeting
the CNS vasculature, and the systemic vasculitides which may affect the CNS
among other organs and systems. Questi includono angioite primaria del
sistema nervoso centrale (PACNS), una malattia rara con destinatario principale
la vascolarizzazione del SNC, e le vasculiti sistemiche che possono influenzare
il sistema nervoso centrale tra gli altri organi e sistemi. Both situations are severe and convey a guarded prognosis.
Entrambe le situazioni sono gravi e trasmettono una prognosi riservata. PACNS usually presents with headache and cognitive
impairment. PACNS di solito si presenta con mal di testa e
deterioramento cognitivo. Focal symptoms are
infrequent at disease onset but are common in more advanced stages. I
sintomi focali sono frequenti all'esordio della malattia, ma sono comuni nelle
fasi più avanzate. The diagnosis of PACNS is
difficult because, although magnetic resonance imaging is almost invariably
abnormal, findings are non specific. La diagnosi di PACNS è difficile
perché, anche se la risonanza magnetica è quasi sempre anormale, i risultati
non sono specifici. Angiography has limited
sensitivity and specificity. L'angiografia ha una sensibilità e
specificità limitata. Brain and leptomeningeal
biopsy may provide a definitive diagnosis when disclosing blood vessel inflammation
and are also useful to exclude other conditions presenting with similar
findings. Cervello e la biopsia leptomeningea possono fornire una
diagnosi definitiva nel comunicare infiammazione dei vasi sanguigni e sono
utili anche per escludere altre condizioni che presentano risultati simili. However, since lesions are segmental, a normal biopsy does
not completely exclude PACNS. Tuttavia, poiché le lesioni sono
segmentale, una biopsia normale non esclude completamente la PACNS. Secondary CNS involvement by systemic vasculitis occurs in
less than one fifth of patients but may be devastating. Coinvolgimento
del SNC secondaria da vasculite sistemica si verifica in meno di un quinto dei
pazienti, ma può essere devastante. A prompt
recognition and aggressive treatment is crucial to avoid permanent damage and
dysfunction. Una tempestiva identificazione e trattamento aggressivo è
fondamentale per evitare danni permanenti e disfunzioni. Glucocorticoids and cyclophosphamide are recommended for
patients with PACNS and for patients with secondary CNS involvement by
small-medium-sized systemic vasculitis. Glucocorticoidi e ciclofosfamide
sono raccomandati per i pazienti con PACNS e per i pazienti con interessamento
del SNC secondario da vasculite sistemica piccole-medie dimensioni. CNS involvement in large-vessel vasculitis is usually
managed with high-dose glucocorticoids (giant-cell arteritis) or
glucocorticoids and immunosuppressive agents (Takayasu's disease).
Coinvolgimento del SNC in grandi vasi vasculite è di solito gestito con
glucocorticoidi ad alte dosi (arterite temporale) o glucocorticoidi e agenti
immunosoppressori (malattia di Takayasu). However,
in large vessel vasculitis, where CNS symptoms are usually due to involvement
of extracranial arteries (Takayasu's disease) or proximal portions of
intracranial arteries (giant-cell arteritis), revascularization procedures may
also have an important role. Tuttavia, in grande vasculite nave, se i
sintomi a carico del SNC sono di solito a causa del coinvolgimento delle
arterie extracraniche (malattia di Takayasu) o porzioni prossimali delle
arterie intracraniche (arterite temporale), procedure di rivascolarizzazione
possono anche avere un ruolo importante.
Keywords: Vasculitis, Central
nervous system.Parole chiave: vasculite, sistema
nervoso centrale.
Il sistema
nervoso centrale (CNS) può essere coinvolto da una varietà di malattie
infiammatorie dei vasi sanguigni. Questi includono angioite primaria del
sistema nervoso centrale (PACNS), una malattia rara destinatario principale la
vascolarizzazione del SNC, e le vasculiti sistemiche che possono influenzare il
sistema nervoso centrale tra gli altri organi e sistemi. Entrambe le
situazioni sono gravi e trasmettono una prognosi riservata. PACNS solito
si presenta con mal di testa e deterioramento cognitivo. I sintomi focali
sono frequenti all'esordio della malattia, ma sono comuni nelle fasi più
avanzate. La diagnosi di PACNS è difficile perché, anche se la risonanza
magnetica è quasi sempre anormale, i risultati non sono specifici.L'angiografia
ha una sensibilità e specificità limitata. Cervello e la biopsia
leptomeningea possono fornire una diagnosi definitiva nel comunicare
infiammazione dei vasi sanguigni e sono utili anche per escludere altre condizioni
che presentano risultati simili. Tuttavia, poiché lesioni sono segmentale,
una biopsia normale non esclude completamente PACNS. Coinvolgimento del
SNC secondaria da vasculite sistemica si verifica in meno di un quinto dei
pazienti, ma può essere devastante. Una tempestiva identificazione e
trattamento aggressivo è fondamentale per evitare danni permanenti e
disfunzioni. Glucocorticoidi e ciclofosfamide sono raccomandati per i
pazienti con PACNS e per i pazienti con interessamento del SNC secondario da vasculite
sistemica piccole-medie dimensioni. Coinvolgimento del SNC in grandi vasi
vasculite è di solito gestito con glucocorticoidi ad alte dosi (arterite
temporale) o glucocorticoidi e agenti immunosoppressori (malattia di
Takayasu). Tuttavia, in grande vasculite nave, se i sintomi a carico del
SNC sono di solito a causa del coinvolgimento delle arterie extracraniche
(malattia di Takayasu) o porzioni prossimali delle arterie intracraniche
(arterite temporale), procedure di rivascolarizzazione possono anche avere un
ruolo importante.
Parole chiave: vasculite,
sistema nervoso centrale.
1. INTRODUZIONE
Il
sistema nervoso (CNS) vascolare centrale può essere preso di mira da un gruppo
eterogeneo di malattie infiammatorie. Nella sua forma primaria isolata , l’angioite
del SNC (PACNS) è una rara forma di vasculite ad eziologia sconosciuta, che
colpisce principalmente i vasi piccoli e medi che riforniscono il parenchima
cerebrale, midollo spinale e leptomeningi [1 - 3]. PACNS si traduce in segni
e sintomi di disfunzione del sistema nervoso centrale con clinicamente
apparente partecipazione di altri organi. Il sistema nervoso centrale può
anche essere indirizzata, tra gli altri territori, da vasculiti
sistemiche [4,5]. Questa recensione si
concentrerà su aspetti diagnostici e terapeutici di PACNS e il coinvolgimento
del sistema nervoso centrale da vasculiti sistemiche secondario in età
adulta. Primaria e secondaria del sistema nervoso centrale vasculite
durante l'infanzia sono stati affrontati in ottime recensioni [6 - 8].
2. VASCULITE PRIMARIA del SCN
2.1. Epidemiologia
Data la
rarità PACNS e l'assenza di test diagnostici definitivi, studi epidemiologici
sono praticamente inesistenti. Un'incidenza annuale di 2,4 per milione di
persone è stato recentemente stimato in America del Nord [9]. PACNS è stata riportata
nei bambini [6 - 8] e negli
anziani. Tuttavia, sembra essere più frequente nei maschi nei loro quarta
e quinta decade di vita [2,9]. PACNS può rappresentare
1,2% di vasculite coinvolge il CNS [3].
2.2. Patogenesi
La
patogenesi della PACNS è sconosciuta. Simile ad altre malattie
infiammatorie o autoimmuni croniche, PACNS è pensato per essere innescato da
infezione. Citomegalovirus, virus Ebstein-Barr, virus varicella-zoster, il
virus di immunodeficienza umana, micoplasma e clamidia sono stati considerati
data la capacità di questi agenti per la produzione di lesioni
vasculitiche [10 - 15]. Tuttavia, nella maggior
parte dei pazienti con PACNS non può essere dimostrato un potenziale rapporto
con questi o altri agenti infettivi.
La
natura granulomatosa delle lesioni infiammatorie vascolari nella maggioranza dei
casi suggerisce una risposta Th1-mediata [3,16]. Citochine Th1-correlati
possono promuovere l'infiammazione vascolare in PACNS come suggerito da diversi
modelli sperimentali. Iniezioni intracerebrale di interferone-gamma hanno
dimostrato di innescare lesioni infiammatorie e vasculite nei ratti. [17]. Fattore di necrosi
tumorale (TNF) e dell'interleuchina-6 funzioni proinfiammatorie possono anche
contribuire a infiammazione vascolare in PACNS [18,19]. TNF / recettore TNF topi
transgenici p75 sviluppano multifocale CNS danno ischemico secondario a
vasculite [18]. Elevata CSF IL-6 è stato
trovato in 3 pazienti con diversi tipi di vasculite (poliartrite nodosa,
arterite temporale e la malattia di Behçet) che coinvolgono il sistema nervoso
centrale[19]. Le attuali conoscenze
della fisiopatologia della PACNS è un progresso molto limitato ritardo nella
diagnosi e nella gestione dei pazienti affetti.
2.3. Patologia
PACNS
genere comporta arterie di dimensioni medio-piccole e vene, soprattutto quelle
situate in leptomeningi e aree sottocorticali. Le caratteristiche
alterazioni anatomopatologiche sono costituiti da infiltrazione infiammatoria
delle pareti dei vasi dai linfociti T e macrofagi attivati che subiscono
differenziazione granulomatosa con la formazione a cellule giganti [3,16]. Cellule infiammatorie
infiltrano l'avventizia e successivamente progredire attraverso la parete
arteriosa che causa la frammentazione della lamina elastica
interna. Proliferazione intimale e fibrosi che porta a occlusione vascolare
è spesso osservata[3,16] (Fig. 11). Questo granulomatosa modello è il più comunemente visto e ha
portato alla precedentemente utilizzato granulomatosa termine angiitis del
SNC [3,16,20]. Tuttavia, le
caratteristiche granulomatose non possono essere sempre rispettati e alcuni
esemplari rivelare il cosiddetto atipico CNS angioite modelli che consiste in
infiltrati in prevalenza linfocitaria (modello linfocitica), vasculite
necrotizzante con necrosi fibrinoide (modello necrotizzante) o modelli
misti [20]. In alcuni casi, linfociti
B e plasmacellule possono essere osservati anche [21]. Vascolare β depositi
di amiloide può essere trovata in un sottogruppo di pazienti[20].
Molteplici non specifici, lesioni iperintense in T2, un paziente
di 63- anni con sospetta angioite primaria del sistema nervoso centrale che ha
presentato con mal di testa e deterioramento cognitivo. B) modello
granulomatosa di angioite primaria del sistema nervoso centrale.Transmurale ...
Sebbene
la maggior parte dei pazienti con PACNS presentano principalmente a disfunzioni
del sistema nervoso centrale, gli studi della necroscopia possono divulgare
vasculite clinicamente asintomatica in altre località, tra cui i polmoni, i reni
e del tratto gastrointestinale [3,5,16]. Distinzione da vasculite
sistemica con interessamento del SNC prominente può essere a volte difficile da
stabilire.
2.4. Manifestazioni
cliniche
A seconda delle aree del
cervello coinvolte, PACNS può comprendere un'ampia varietà di dati
clinici.Inoltre, la gravità della malattia e la rapidità di progressione può
essere molto variabile tra i pazienti, aumentando eterogeneità nella
presentazione clinica.
Nel più
grande serie segnalate tra cui 101 pazienti [9], l'età media alla diagnosi
era di 47 anni (range 17-84 anni). La maggioranza dei pazienti ha
presentato con manifestazioni subacute di disfunzione del sistema nervoso
centrale diffusa. Presentazione acuta era altamente insolito. I primi
sintomi più comuni sono stati: cefalea (63%) e deficit cognitivo
(50%). Mal di testa erano inizialmente di bassa intensità e progressivamente
peggiorato. Il deficit cognitivo è stato anche insidioso. Sintomi
focali generalmente apparivano più avanti nel corso della malattia e
includevano emiparesi (44%), ictus (40%), l'afasia (28%), attacco ischemico
transitorio (28%), atassia (19%), convulsioni (16%) , disartria (15%) e
offuscamento della vista o acuità visiva ridotta (11%). Manifestazioni
frequenti, si verificano in meno del 10% dei pazienti, inclusa emorragia
intracranica, la sindrome amnesico, manifestazioni del midollo spinale, come paraparesi
o tetraparesi, parkinsonismo, vertigini, capogiri o paralisi del nervo
cranico. La maggior parte dei pazienti avevano molteplicimanifestazioni. Altri
Report serie pubblicata reperti simili [22,23].
Al fine
di facilitare il riconoscimento clinico e la diagnosi precoce, le
manifestazioni cliniche sono stati raggruppati in tre principali fenotipi: 1)
acuta o più comunemente encefalopatia subacuta, presentandosi come una sindrome
confusionale con la progressione di stupore e coma; 2) presentazione della
malattia simile atipica sclerosi multipla con una varietà di sintomi focali
come la neuropatia ottica, episodi del tronco cerebrale, crisi epilettiche, mal
di testa, gli episodi encefalopatici o eventi simil-ictus emisferici e 3) le
lesioni di massa intracraniche, con mal di testa, sonnolenza, segni focali e
pressione intracranica elevata [24,25].
È stato
anche suggerito che il coinvolgimento predominante rispetto piccola
imbarcazione medie può influenzare presentazione della malattia. Piccoli
vasi PACNS manifesta come una subacuta o encefalopatia acuta con mal di testa
persistente, deficit cognitivo, confusione e convulsioni. MRI solito
rivela marcato miglioramento del contrasto meningeo mentre l'angiografia non
può rivelare cambiamenti perché i vasi interessati sono piccole, al di là della
soglia di rilevamento [26,27]. Questa forma di PACNS può
rispondere alla monoterapia con glucocorticoidi, ma il 25% dei pazienti
recidiva. Al contrario, quando si tratta di navi di medie dimensioni, in
aggiunta al mal di testa e disfunzioni generale CNS, deficit neurologici focali
e ictus sono più comuni e angiografia è più probabile rivelare anomalie
vascolari [9,26,27]. Quattro caratteristiche
cliniche sono associate ad un aumento della mortalità nei pazienti con PACNS:
deficit neurologico focale, deterioramento cognitivo, infarto cerebrale e il
coinvolgimento di navi più grandi [9].
I
sintomi generali e risultati che suggeriscono una certa misura di
coinvolgimento sistemico si possono verificare. Febbre, perdita di
peso, livedo reticularis, rash, neuropatia periferica, artrite
e sudorazione notturna può essere registrata nel 20% dei pazienti [2,9].
2.5. Diagnosi
La
diagnosi di PACNS è una sfida a causa della mancanza di test diagnostici
altamente sensibili e specifici. ,, Neuroimaging e dati istopatologici
analitici clinici sono importanti, sia nel sostenere il sospetto diagnostico e
ad escludere altre condizioni che si presentano con caratteristiche simili.
2.5.1. Anomalie di laboratorio di prova
Gli
esami di routine sono spesso all'interno del range di normalità [2,9,28]. In alcune caratteristiche
dei pazienti di risposta infiammatoria sistemica tra cui anemia, leucocitosi e
moderato aumento delle proteine di fase acuta può essere osservato (VES, proteina C-reattiva e la conta
piastrinica)[2,9]. Gli esami di laboratorio
sono utili per escludere altre malattie che possono presentare sintomi simili
come infezioni, vasculite sistemica, malignità, l'abuso di droga e gli stati
ipercoagulabilità [5,28,29].
Liquido
cerebrospinale (CSF) è anormale nel 80-90% dei pazienti [9]. Una maggiore
concentrazione di proteine è il reperto più comune.In una serie di 101 pazienti, significa che la concentrazione di
proteine CSF era
7 gr / L (range 1,5-10,3 gr / L)[9]. La pressione viene
aumentata nel 50% dei pazienti e conta dei linfociti elevati può osservare nel
50-80%. CSF immunoglobuline oligoclonali possono essere trovati nel 50%
delle persone fisiche con PACNS [5,23]. CSF pleiocitosi è
modesto, raramente superiore a 250 cellule / ml.Elevati conta dei leucociti e
la presenza di neutrofili sono rari e, quando presenti, devono segnalare
l'eventuale infezione [2]. Analisi CSF è utile per
escludere infezioni e neoplasie maligne e appropriate macchie batteriche e
fungine, reazioni a catena della polimerasi virale, e citometria a flusso studi
dovrebbero essere eseguiti.
2.5.2. Imaging
2.5.2.1. Risonanza
Magnetica (MRI) e Risonanza Magnetica Angiografia (MRA)
RM è
sensibile ma non specifico nel rivelare cambiamenti associati con PACNS [30]. Le lesioni sono spesso molteplici
e bilaterale e comprendono parenchimali o zone d'aumento meningea, aree
ischemiche o infarti nella corteccia, la materia bianca profonda, o
periventricolare sostanza bianca (Fig. 1A1A). Essa può anche rivelare lesioni emorragiche [31,32]. La sensibilità della RM
in PACNS confermato da biopsia è molto alto, rivelare anomalie nel 97% dei
casi [22,32 - 34] ma risultati anomali sono
non specifici. Imaging ponderata diffusione è altamente sensibile nel
rilevare anomalie di diffusione e può essere utile nei pazienti con normale
risonanza magnetica [35]. MRA ha una sensibilità
limitata ed è solo in grado di rivelare anomalie nei grandi vasi
intracranici. Le stesse limitazioni valgono per CT-angiografia [33,34].
2.5.2.2. Angiografia
Convenzionale
Angiografia
convenzionale è la tecnica di imaging più specifico per la diagnosi di PACNS e,
rispetto a MRA è in grado di rilevare anomalie in vasi
piccoli. Caratteristiche tipiche angiografici di PACNS sono più
"perline" o segmentale restringimento in grandi, intermedie o piccole
arterie con le regioni interposte di ectasia o di architettura normale del
lume [31 - 33] (Fig. 1D1D). Perline può essere liscia o irregolare e verifica in genere
bilaterale. Ulteriori modifiche sono aneurismi, flusso collaterale, aree
isolate di navi restringimento in più rami, irregolarità dei vasi circolari o
eccentrici, più occlusioni con tagli netti e le lesioni di massa apparentemente
avascolari [31 - 33].
Anche
se i risultati di sistema nervoso centrale angiografie convenzionali possono
supportare la diagnosi di PACNS e può essere utilizzato per dirigere il sito di
biopsia, nessuno di questi risultati non è solo diagnostico perché le immagini
simili possono essere presenti in altre malattie (tabelle 11 e 22) [ 2,5,22,28,36 - 38].
Clinica, Laboratorio, per immagini e istopatologiche
caratteristiche utile distinguere RVCS da PACNS
Anche
se essenziale per la diagnosi, l'angiografia ha limitata sensibilità e
specificità. I pazienti con PACNS confermato da biopsia possono avere
normali angiografie che appaiono e, viceversa, le biopsie di vasi anomali
angiograficamente sono stati segnalati come normale [2,5,28]. La sensibilità
dell'angiografia nel rilevare PACNS varia dal 20% al 90% [1,9,31,35,37,38] e specificità dal 20 al
60% [1,9,31,34]. La sensibilità di
angiografia cerebrale diminuisce con il calibro dei vasi coinvolti, essendo più
sensibile per il coinvolgimento di navi di grandi e medie
dimensioni. L'angiografia non è esente di effetti collaterali. Circa
0,8% dei pazienti sottoposti a esperienza angiografia deficit neurologici
aggiuntivi come evento avverso correlato alla procedura [32]. Tuttavia, data la gravità
della PACNS e la difficoltà di raggiungere una diagnosi accurata, il rapporto
rischio / beneficio è accettabile e angiografia convenzionale è raccomandato
come procedura diagnostica chiave.
2.5.2.3. Esame istopatologico
Biopsia
cerebrale è considerata il gold standard per la diagnosi di PACNS ma rivela
anomalie istopatologiche diagnostici solo nel 50% al 75% dei casi [1] (Fig. 1B1B e CC). Il ruolo della biopsia cerebrale in PACNS non è limitata a
dimostrare l'infiammazione dei vasi sanguigni: è anche importante escludere
altre condizioni come infezioni, neoplasie, malattie degenerative o per le
quali sono richieste completamente differenti approcci di trattamento
(Tabella 11)[5 , 27].
Nel più
grande serie di pazienti sottoposti a biopsia chirurgica PACNS, tra cui 43
pazienti, sensibilità diagnostica della biopsia cerebrale è stata del 63% [20]. In questa serie, la
distribuzione dei vari modelli morfologici era la seguente: necrotizzante acuta
(14%), puramente linfocitica (28%) e granulomatosa (58%), senza differenze
statisticamente significative in aggressività della malattia o la risposta al
trattamento tra loro.È interessante notare che il 78% delle biopsie dirette ad
una anomalia per immagini erano diagnostico, mentre nessuno dei biopsie cieche
dimostrato vasculite. Le biopsie comprese leptomeningi erano leggermente
più sensibile nel rilevare vasculite quelli non compresi esso (58% contro
40%). In accordo con questi risultati altri autori hanno riportato una
sensibilità di biopsia cerebrale circa il 50% [2,16]. L'alta percentuale di
biopsie negative nei pazienti con caratteristiche cliniche e radiografiche di
grande suggestione di PACNS può essere spiegato dalla natura segmentale delle
lesioni. Inoltre biopsie sono di solito presi dal parenchima superficiale
e leptomeningi e, in alcuni casi, vasi coinvolti sono di dimensioni maggiori e
si trovano più in profondità da queste aree [20]. Per massimizzare la
sensibilità diagnostica della procedura è consigliabile biopsie vengono
eseguite in zone anomale rilevate da immagini precedenti e includono
leptomeningi. Biopsia stereotassica è raccomandato per le lesioni di massa
solo [20,25].
Occasionalmente,
depositi di amiloide possono osservare [20,25]. Questi sono più
frequentemente trovati in campioni con un modello granulomatosa e coloro che le
presentano come lesioni di massa [20,25].Clinicamente, i pazienti con
depositi di amiloide sono più vecchi e più frequentemente si presentano con
esordio acuto e deterioramento cognitivo [39]. Esito clinico e risposta
al trattamento sembra essere simile a quella dei pazienti senza depositi
amiloidi [39].
2.5.2.4. Criteri diagnostici
Dal
momento che la conferma istopatologica di PACNS non è sempre fattibile,
Calabrese e Mallek hanno proposto una serie di criteri diagnostici combinando,
clinica, imaging e istopatologici [1]. Questi includono: 1)
deficit neurologico che rimane inspiegata dopo un percorso diagnostico
vigoroso, compresi gli studi di puntura lombare e di neuroimaging, 2) le
anomalie angiografiche altamente suggestivi di vasculite o evidenza
istopatologica di vasculite all'interno del sistema nervoso centrale e 3)
nessuna evidenza di vasculite sistemica o di qualsiasi altro condizione alla
quale i rilievi angiografici o patologici possono essere
attribuiti. Queste condizioni sono riportate nella Tabella 11 (Fig. 2 2).
Puntiforme T2 iperintense in lesioni della sostanza bianca una
donna di 40- anni con la sindrome di Susac. Questo paziente aveva anche
ipoacusia neurosensoriale e della retina occlusione bilaterali ramo
dell'arteria come parte della sindrome.
2.5.2.5. Trattamento
Nessun
studi randomizzati controllati o studi prospettici sono stati condotti con
pazienti affetti da PACNS.Pertanto, le raccomandazioni terapeutiche si basano
su estrapolazioni di dati ottenuti da studi condotti in altre vasculiti
sistemiche gravi, studi retrospettivi, piccola serie caso e opinion di
esperti [2,5,40]. In una rassegna retrospettiva
dei trattamenti ricevuti da 101 pazienti con diagnosi di PACNS (70 per
angiografia, 31 da biopsia) Salvarani et al., Hanno trovato
che 97 pazienti sono stati trattati con glucocorticoidi, 25 delle quali con
impulsi metil-prednisolone per via endovenosa 1gr e per il restante con orale
prednisone alla dose media di 60 mg / giorno [9]. Quarantanove pazienti
hanno ricevuto un agente immunosoppressivo: 46 ciclofosfamide (per via orale a
150 mg / die o endovenosa di circa 1 gr / mese) e azatioprina 3. Una
risposta favorevole è stata osservata nel 81% dei pazienti trattati con i soli
glucocorticoidi e nel 81% di quelli trattati con entrambi prednisone e
ciclofosfamide. Data la natura retrospettiva dell'indagine non è possibile
concludere che gli agenti immunosoppressivi non sono necessari poiché il gruppo
trattato con ciclofosfamide può essere considerata più grave trattando medici.
Il
trattamento con glucocorticoidi (prednisone orale o equivalente a 60 mg / die
preceduto da tre 1 gr impulsi per via endovenosa nei casi più gravi) dovrebbero,
quindi, essere avviati al più presto CNS vasculite (primaria o secondaria) è
clinicamente sospetta e malattie infettive ragionevolmente
esclusa. Prednisone può essere rapidamente rastremato se la diagnosi è
infine esclusa. Quando la diagnosi di vasculite sistema nervoso centrale è
sostenuto anche da angiografia o la biopsia e imita sono esclusi in modo
convincente, si consiglia di ciclofosfamide (per via orale di 150 mg / die o
impulso mensile 1gr). Pulse ciclofosfamide per via endovenosa ha un'efficacia
equivalente a indurre la remissione, ma è meno tossico giornaliero
ciclofosfamide per via orale a vasculite sistemica [40]. Per analogia a grave
vasculite sistemica, passare a un agente immunosoppressivo più sicuro
(azatioprina, metotrexato o micofenolato) possono essere considerati dopo 4-6
mesi di trattamento con ciclofosfamide [il 40 - 43]. Tutti i pazienti
dovrebbero essere dati calcio e vitamina D, agenti di protezione delle ossa e
la profilassi delle infezioni Pneumocystis[5].
Recentemente
è stato dimostrato che Rituximab è altrettanto efficace di ciclofosfamide
nell'indurre remissione nella vasculite sistemica ANCA-associata grave [44,45]. Rituximab è stato anche
successo nel trattamento di pazienti affetti da LES con interessamento del
SNC [46], ma non vi è alcuna
esperienza con rituximab in PACNS. Due glucocorticoidi e casi refrattari
ciclofosfamide rispondono al TNF blocco sono stati riportati[47].
Trattamento
Immunossuppressive deve essere mantenuta per 2-3 anni [2,5]. E 'importante tenere a
mente che circa il 25% dei pazienti può recidiva [9]. La risposta al
trattamento deve essere monitorata da una valutazione neurologica periodica e
esame MRI seriale ogni 3-4 mesi [2,28].
RCVS è
un termine recentemente proposto per descrivere il substrato fisiopatologico di
un gruppo di condizioni caratterizzate da prolungata ma reversibile
vasocostrizione delle arterie cerebrali [48]. In precedenza, queste
sindromi sono stati denominati come angiopatia benigna del sistema nervoso
centrale e, per molti anni, non vi è stata una netta distinzione tra RCVS e
vero angioite primaria del sistema nervoso centrale. RCVS ha ricevuto una
varietà di nomi: sindrome di Call-Fleming, rombo di tuono mal di testa con
vasospasmo reversibile, vasospasmo emicranica o emicrania angioite, dopo il
parto angiopathy, o farmaco-indotta arterite cerebrale o angiopatia [48].
RCVS
può avvenire spontaneamente, ma nella maggior parte dei casi è associato a
fattori scatenanti, tra cui l'uso di sostanze vasoattive (cioè derivati ergotamina, anfetamine e
decongestionanti nasali) altri farmaci (ad esempio gli inibitori della
ricaptazione della serotonina selettivi, contraccettivi), droghe ricreative
(cannabis, ecstasy, LSD , cocaina, alcol), la gravidanza o puerperio in
ritardo, il rapporto sessuale, e la produzione di catecolamine tumori [48 - 50]. Il più caratteristico
manifestazione clinica iniziale includono iperacuto forte mal di testa e
ricorrenti che possono essere associati con sintomi e segni neurologici [48]. Mal di testa di solito è
diffusa anche se può essere anche localizzata, preferenzialmente nella zona
occipitale, e può essere associata a nausea, vomito e
fotosensibilità. Altre manifestazioni cliniche comprendono disfunzione
visiva, attacchi ischemici transitori e sequestri [48]. La principale
complicazione di RCVS è colpo che può portare alla sequele permanenti e anche
la morte [48,49]. Sebbene la fisiopatologia
di RCVS non è noto, l'ipotesi prevalente ritiene che esista un disturbo
transitorio nel controllo del tono vascolare cerebrale [48].
Nel più
grande serie segnalate tra cui 67 pazienti [49], c'era una predominanza
femminile (67%) con un'età media alla diagnosi di 42,5 ± 11,8 anni (range 19-70
anni). Fattori precipitanti sono stati identificati nel 63%, essendo l'uso
di sostanze vasoattive più frequente (55%). Il sintomo che presenta in
tutti i casi, è stato recente forte mal di testa, e questo era l'unico sintomo
nel 76%. Tra i 67 pazienti, il 94% ha più mal di testa Thunderclap (media
di 4,5 episodi) che ricorreva nel corso di un periodo medio di 1
settimana. In questa serie, complicanze precoci (entro la prima settimana)
inclusa emorragia subaracnoidea corticale (22%), della leucoencefalopatia
posteriore reversibile (9%), intrecerebral sanguinamento (6%) e sequestri
(3%).Complicazioni ritardate (dopo la prima settimana) incluso attacco
ischemico transitorio nel 16% e infarti cerebrali a 4%. Il risultato complessivo
di questa serie è stato buono, senza ricadute nel corso di un periodo di
follow-up 16 ± 12,4 mesi e solo il 4% dei pazienti ha avuto deficit neurologici
persistenti.
In
assenza di criteri diagnostici convalidati, Calabrese et al.[48] proposto una serie di
elementi chiave necessari per la diagnosi di RCVS. Questi includono gravi,
mal di testa acuti, con o senza ulteriori segni neurologici o sintomi, normale
o vicino al normale analisi del liquido cerebrospinale, test di neuroimaging
(angiografia transfemorale, angio-TC o MRA) documentano segmentale multifocale
cerebrale vasocostrizione, senza evidenza di emorragia subaracnoidea
aneurismatica , e la reversibilità delle anomalie angiografiche entro 12
settimane [47 - 49]. Il trattamento consiste
solitamente di calcio-antagonisti [48 - 51] e brevi corsi di
glucocorticoidi [50,52].
La
distinzione di PACNS e RVCS è importante a causa dei diversi requisiti di
prognosi e il trattamento.Elementi chiave per distinzione sono stati
proposti [2,48] e sono riassunti nella
tabella 22.PACNS
colpisce in genere uomini di mezza età, mentre RVCS è principalmente una
malattia di donne tra i 20-40 anni. In quest'ultimo quasi il 60% dei
pazienti segnalare un evento precipitante [48], di solito esposizione a
sostanze vasoattive. Mal di testa in PACNS è indolente e progressiva [9] che, mal di testa in RVCS
è acuta e grave [2,48,49]. A meno complicata da
emorragie o infarto, la risonanza magnetica non rivela importanti cambiamenti
nel RVCS mentre la RM è anormale nel 97% dei casi con PACNS [9,50]. Per definizione, anomalie
angiografiche sostanzialmente o completamente reversibili entro circa 3 mesi.
4. vasculiti SISTEMICHE COINVOLGE LA CNS
La
vascolarizzazione del SNC può essere bersaglio di vasculite sistemica (Tabella 3 3). Di solito coinvolgimento del SNC coesiste con altre
manifestazioni sistemiche chiaramente evidenti ma alcuni pazienti può
presentare soprattutto con i sintomi prominenti di CNS disfunzione [4,5,53]. In vasculite sistemica
mirato ai vasi di piccole-medie dimensioni, il coinvolgimento del sistema
nervoso centrale è un predittore di prognosi sfavorevole / custodito [54,55] ed è uno dei fattori considerati
per consigliare il trattamento aggressivo con ciclofosfamide, oltre a alte dosi
di steroidi [40,54,55]. Tuttavia, in grandi vasi
vasculite, il coinvolgimento del sistema nervoso centrale può beneficiare di
procedure vascolari di intervento (angioplastica, un intervento chirurgico
derivativo), antiaggreganti o trattamento anticoagulante in aggiunta ai
glucocorticoidi alte dosi piuttosto che intensificazione della terapia
immunosoppressiva [56 - 58].
Primaria sistemica vasculite Coinvolgere più di frequente del
sistema nervoso centrale negli adulti
4.1. CNS coinvolgimento da parte delle piccole e medie
Vessel Sized Vasculitis
A
livello globale, il coinvolgimento cerebrospinale non è frequente nelle
piccole-medie dimensioni vasculite nave, tra cui granulomatosi di Wegener,
poliangioite microscopica, sindrome di Churg-Strauss, poliarterite nodosa,
vasculite crioglobulinemica, e la malattia di Behçet. Coinvolgimento del
SNC si verifica in meno del 15% dei pazienti nella maggior serie.
4.1.1. Wegener Granulomatosis (WG) Granulomatosi di
Wegener (WG)
The prevalence of CNS manifestations in WG ranges from 2.7% to 9%
in large series of patients [ 59 - 61 ]. La prevalenza delle manifestazioni del sistema nervoso centrale
in WG varia dal 2,7% al 9% in grande serie di pazienti [59 - 61].Neurological
involvement may account through 3 major mechanisms: vasculitis involving CNS
vessels, granulomatous lesions located in the brain, meninges or cranial nerves
and direct extension of destructive granulomatous tissue from nasal or
paranasal structures [ 59 - 62 ]. Coinvolgimento neurologico può spiegare attraverso 3 principali
meccanismi: vasculite che coinvolge i vasi del sistema nervoso centrale,
lesioni granulomatose situati nel cervello, meningi o nervi cranici e diretta
estensione del tessuto granulomatoso distruttivo da nasale o strutture paranasali
[59 - 62].
Cerebral vasculitis is the most frequent CNS lesion and may
present with headache, visual disturbances, seizures, confusion, ischemic
stroke, intracerebral or subarachnoid haemorrhage, venous thrombosis or
dementia [ 62 , 63 ]. Vasculite cerebrale è il più frequente del sistema nervoso
centrale lesione e può presentare con mal di testa, disturbi visivi,
convulsioni, confusione, ictus ischemico, intracerebrale o emorragia
subaracnoidea, trombosi venosa o demenza [62,63].Granulomatous
inflammation and thickening of the duramater, pachymeningitis, may present with
chronic headache, multiple cranial nerve palsies, seizures, meningeal signs,
encephalopathy, proptosis, limb palsy or ataxia [ 62 - 65 ]. Infiammazione granulomatosa e l'ispessimento della dura madre,
pachimeningite, possono presentarsi con cefalea cronica, più paralisi dei nervi
cranici, convulsioni, segni meningei, encefalopatia, proptosi, arto paralisi o
atassia [62 - 65].Pituitary
involvement leads to central diabetes insipidus, panhypopituitarism or a
combination of hormone deficiencies [ 66 ]. Coinvolgimento pituitaria porta a diabete insipido centrale,
panipopituitarismo o una combinazione di carenze ormonali [66].In
these patients, MRI is the image technique of choice because it can reveal
ischemic or hemorrhagic lesions, dural thickening, pituitary involvement or
enhancement of inflamed orbital and paranasal mucosa [ 63 ]. In questi pazienti, la RM è la tecnica immagine di scelta, perché
può rivelare lesioni ischemiche o emorragiche, ispessimento durale,
coinvolgimento ipofisi o il potenziamento di infiammata orbitale e paranasali
mucosa [63].In
the case of dural involvement, tissue biopsy may disclose granulomatous
pachymeningitis [ 66 ]. Nel caso di coinvolgimento durale, la biopsia del tessuto può
rivelare pachimeningite granulomatosa [66].
In a series of 85 patients, CNS involvement was present in 10
cases (11.8%) and CNS vasculitis was the cause of death of one of them [ 67 ]. In una serie di 85 pazienti, il coinvolgimento del sistema
nervoso centrale era presente in 10 casi (11,8%) e del sistema nervoso centrale
vasculite è stata la causa della morte di uno di essi [67].
There are only scattered case reports of CNS manifestations
related to MPA in the literature. Ci sono
segnalazioni di casi solo sparsi di manifestazioni del sistema nervoso centrale
relativi al MPA in letteratura. Multiple bilateral
cerebral infarctions [ 68 ], multiple
hemorrhagic infarction of the cerebral cortex caused by CNS vasculitis [ 69 ], capsular
warning syndrome and subsequent stroke [ 70 ] and
pachymeningitis have been occasionally reported [ 71 , 72 ]. Molteplici infarti cerebrali bilaterali [68], più emorragico infarto della
corteccia cerebrale causata da CNS vasculite [69], la sindrome di avvertimento
capsulare e la successiva corsa [70] e pachimeningite sono state
riscontrate occasionalmente [71,72].
4.1.3. 4.1.3. Churg-Strauss Syndrome (CSS) Sindrome di
Churg-Strauss (CSS)
In the largest published series of CSS patients the CNS is
reported to be involved in 8% to 14% of patients [ 73 - 77 ]. Nel più grande serie pubblicata di pazienti CSS SNC è segnalato
per essere coinvolti in l'8% al 14% dei pazienti [73 - 77].
Cerebral infarction is the most frequently reported manifestation
of CNS involvement [ 75 , 77 ], probably
as result of cerebral vasculitis (Fig. 3 3
). Infarto cerebrale è la manifestazione più frequentemente riferito
coinvolgimento del SNC [75,77], probabilmente come risultato
di vasculite cerebrale (Fig. 3 3).Additional less commonly reported CNS events include
intracerebral haemorrhage [ 78 , 79 ] and
pachymeningitis [ 80 , 81 ]. Ulteriori eventi a carico del SNC meno comunemente riportati
includono emorragia intracerebrale [78,79] e pachimeningite [80,81].
Multiple brain infarcts in a patient with Churg-Strauss syndrome.
B ) CT scan from the same patient disclosing pulmonary infiltrates and
bilateral pleural effusion. Molteplici infarti
cerebrali in un paziente con B Churg-Strauss sindrome.) TAC dallo stesso
paziente divulgare infiltrati polmonari e versamento pleurico bilaterale. Toracocentesis disclosed predominance of eosinophils in
pleural fluid exudate. Toracocentesis divulgate predominanza di
eosinofili nel pleurico essudato liquido.
In a recent series of 348 patients diagnosed with PAN over a
42-year period, 4.6% presented with central nervous system-related
abnormalities [ 82 ]. In una recente serie di 348 pazienti con diagnosi di PAN per un
periodo di 42 anni, il 4,6% ha presentato con le anomalie nervoso centrale
relativi al sistema [82].Earlier
studies reported a higher prevalence, between 15 and 65% [ 83 ]. Studi precedenti hanno riportato una più alta prevalenza, tra il
15 e il 65% [83].Perhaps
in present days, earlier recognition of the disease with prompt treatment
prevents development of severe complications. Forse in giorni di
presenza, in precedenza il riconoscimento della malattia con un trattamento immediato
previene lo sviluppo di gravi complicazioni. It is
important to remark that, widespread ANCA and cryoglobulin testing has led to
re-classification of a substantial proportion of patients with necrotizing
vasculitis previously diagnosed with PAN, which, in fact, has become a much
more infrequent disease [ 84 ]. È importante notare che, diffusa test ANCA e crioglobuline ha
portato a ri-classificazione di una proporzione sostanziale di pazienti con
vasculite necrotizzante precedente diagnosi di PAN, che, di fatto, è diventato
una malattia molto più rari [84].
In an extensive literature review, three major clinical
presentations related to CNS involvement have been recognized in PAN: 1)
diffuse encephalopathy characterized by cognitive impairment, disorientation or
psychosis (8% to 20%), 2) seizures (focal or generalized) and 3) focal
neurologic deficits [ 83 ]. In un'ampia revisione della letteratura, tre principali
presentazioni cliniche relative al coinvolgimento del sistema nervoso centrale
sono stati riconosciuti in PAN: 1) encefalopatia diffusa caratterizzata da
deficit cognitivo, disorientamento o psicosi (8% al 20%), 2), convulsioni
(focali o generalizzate) e 3 ) deficit neurologici focali [83].Accelerated
hypertension may also contribute to diffuse encephalopathy in some patients [ 83 ]. Ipertensione accelerata può anche contribuire a diffondere
encefalopatia in alcuni pazienti [83].Abnormal
findings reported in neuroimaging studies (MRI and CTscan) include cerebral
infarctions located in the brain (cortical or subcortical), cerebellum or
brainstem and cerebral hemorrhages [ 85 , 86 ] (Fig. 4 4
). Risultati anomali riportati in studi di neuroimaging (risonanza
magnetica e CTscan) includono infarti cerebrali situati nel cervello (corticale
o sottocorticale), il cervelletto o del tronco cerebrale ed emorragie cerebrali
[85,86] (Fig. 4 4).
Hemorrhagic brain infarct in a patient with systemic poyarteritis
nodosa. Infarto cerebrale emorragica in un paziente con poyarteritis
nodosa sistemica. This patient also had
hypertension, postprandial abdominal pain, multineuritis and livedo
reticularis. B ) Skin biopsy of the same patient disclosing necrotizing
arteritis in the ... Questo paziente aveva anche ipertensione,
dolori addominali postprandiali, multineuritis e livedo reticolare. La biopsia B)
della pelle dello stesso paziente divulga necrotizzante arterite in ...
4.1.5. 4.1.5. Cryoglobulinemia Crioglobulinemia
CNS involvement is uncommon in cryoglobulinemic vasculitis. Coinvolgimento del SNC è raro in vasculite crioglobulinemica. In a retrospective series of 209 patients [ 87 ], CNS
involvement was detected in 3. In a prospective study of 40 patients with mixed
type II cryoglobulinemia vasculitis [ 88 ]
specifically investigating signs of CNS dysfunction, 89% of the patients had
some cognitive impairment, being attention the aspect most commonly altered
(70.3%), followed by alterations in executive functions and visual
construction. In una serie retrospettiva di 209 pazienti
[87], il coinvolgimento del sistema
nervoso centrale è stato rilevato in 3. In uno studio prospettico di 40
pazienti con crioglobulinemia mista di tipo II vasculite [88] in particolare indagando segni
di disfunzione del sistema nervoso centrale, l'89% dei pazienti ha avuto un po
'di deficit cognitivo , essendo l'attenzione l'aspetto più comunemente alterato
(70,3%), seguiti da alterazioni delle funzioni esecutive e costruzione visiva. Whether these abnormalities are due to CNS vasculitis,
co-morbidities, glucocorticoid, immunosuppressive or antiviral treatments or a
combination of factors is unclear. Se queste anomalie sono dovute a CNS
vasculite, co-morbidità, glucocorticoidi, immunosoppressive o trattamenti
antivirali o di una combinazione di fattori è chiaro.
Clinical features of CNS involvement in cryoglobulinemia include
encephalopathy, stroke, transient ischemic attacks, lacunar infarctions and
hemorrhage [ 89 , 90 ]. Le caratteristiche cliniche di coinvolgimento del sistema nervoso
centrale in crioglobulinemia includono encefalopatia, ictus, attacchi ischemici
transitori, infarti lacunari ed emorragia [89,90].Most
of the cases reported are associated to hepatitis C virus infection. La
maggior parte dei casi segnalati sono associati ad infezione da virus
dell'epatite C.
4.1.6. 4.1.6. Behçet's Disease Malattia di Behçet
The frequency of neurological involvement in Behçet's disease
ranges from 5.3% to 14.3% in prospective studies [ 91 , 92 ]. La frequenza di coinvolgimento neurologico nella malattia di
Behçet varia dal 5,3% al 14,3% in studi prospettici [91,92].Neuro-Behçet
occurs more frequently in patients aged 20 to 40 years and is 2-8 times more
frequent in men than in women. Neuro-Behçet si verifica più
frequentemente nei pazienti di età compresa tra i 20 ei 40 anni ed è 2-8 volte
più frequente negli uomini che nelle donne. Neurological
manifestations commonly appear when other systemic features are present.
Manifestazioni neurologiche comunemente appaiono quando sono presenti altre
caratteristiche sistemiche. CNS involvement is the
first disease manifestation in less than 6% of patients with neuro-Behçet [ 93 ]. Coinvolgimento CNS è la prima manifestazione della malattia in
meno del 6% dei pazienti con neuro-Behçet [93].CNS
involvement in Behçet's disease may occur through 2 major mechanisms:
meningoencephalitis and vascular disease. Coinvolgimento del SNC nella
malattia di Behçet può avvenire attraverso 2 meccanismi principali:
meningoencefalite e malattia vascolare.
Meningoencephalitis is usually subacute and predominantly
involves the brainstem but may extend to basal ganglia, thalamus, cortex and
white matter [ 93 , 94 ]. Meningoencefalite è di solito subacuto e coinvolge
prevalentemente il tronco cerebrale, ma può estendersi a gangli basali, talamo,
corteccia e la materia bianca [93,94].The
spinal cord and cranial nerves may also be affected. I nervi del midollo
spinale e cranici possono essere colpiti. In the
largest series of patients with neuro-Behçet [ 92 ] the most
common clinical symptoms were pyramidal signs (96%), hemiparesis (60%),
behavioural changes, headache and sphincter disturbance or impotence. Nel più grande serie di pazienti con neuro-Behçet [92] i sintomi clinici più comuni
sono stati segni piramidali (96%), emiparesi (60%), cambiamenti comportamentali,
mal di testa e disturbi sfintere o impotenza. Less
common manifestations were paraparesis, meningeal signs, movement disorders,
brainstem signs, seizures, hemianopsia, aphasia, psyachiatric disturbances or
cerebellar syndrome. Manifestazioni meno comuni erano paraparesi, segni
meningei, disturbi del movimento, i segni del tronco encefalico, convulsioni,
hemianopsia, afasia, disturbi psyachiatric o sindrome cerebellare. CSF analysis was abnormal 70–80% disclosing moderately
elevated protein concentration and pleocytosis with neutrophilia at early
stages [ 89 ]. Analisi CSF era anormale 70-80% divulgare concentrazione di
proteine moderatamente
elevata e pleiocitosi con neutrofilia nelle fasi iniziali [89].MRI
discloses hyperintense T2 lesions with contrast enhancement and edema.
Risonanza magnetica rivela lesioni T2 iperintense con intensificazione del
contrasto ed edema. Lesions are usually unilateral
and are located in the upper brainstem extending towards the thalamus and basal
ganglia [ 95 ]. Le lesioni sono di solito unilaterale e sono situati nel tronco
cerebrale superiore che si estende verso il talamo e nuclei della base [95].Tumor-like
lesions may occasionally occur [ 93 ]. Lesioni tumorali come possono occasionalmente verificarsi [93].
The most common manifestation of vascular neuro-Behçet is central
venous thrombosis with signs and symptoms of intracranial hypertension,
including papilledema. La manifestazione più comune
di neuro-vascolare Behçet è la trombosi venosa centrale con segni e sintomi di
ipertensione endocranica, compreso papilledema. Intracranial
aneurysms and ischemic stroke may also occur but are infrequent complications.
Aneurismi intracranici e ictus ischemico può anche verificarsi, ma sono
complicazioni frequenti. Combined parenchymal and
vascular involvement may be seen in 20% of patients with neuro-Behçet [ 93 ]. Parenchimale combinato e coinvolgimento vascolare può essere
visto nel 20% dei pazienti con neuro-Behçet [93].Patients
with neuro-Behçet are treated with high-dose glucocorticoids and
cyclophosphamide. I pazienti con neuro-Behçet sono trattati con alte
dosi di glucocorticoidi e ciclofosfamide. Blocking
TNFα with infliximab may be useful in refractory patients. Blocco
TNF con Infliximab può essere utile nei pazienti refrattari.
4.2. 4.2. Large Vessel Vasculitis Vasculite a grandi cellule
Both giant-cell arteritis of the elderly and Takayasu disease may
convey CNS involvement. Entrambi arterite temporale
della malattia anziani e Takayasu può trasmettere il coinvolgimento del sistema
nervoso centrale.
4.2.1. 4.2.1. Giant Cell Arteritis Arterite a cellule giganti
GCA preferentially targets the cranial vessels. GCA rivolge preferenzialmente i vasi cranici. Consequently the most common ischemic complications occur
in territories supplied by the carotid and vertebral arteries. Di
conseguenza, le complicanze ischemiche più comuni si verificano in territori
fornite dal arterie carotidee e vertebrali. Although
GCA is considered a large to medium sized vessel vasculitis, small cranial
vessels are frequently affected [ 96 ] and the
most frequent ischemic complication, visual loss, derives from involvement of
the small arteries supplying the optic nerve [ 97 - 100
]. Sebbene GCA è considerato un grande per dimensioni medie
vasculite nave, piccoli vasi cranici sono frequentemente interessate [96] e la complicanza più frequente
ischemica, perdita della vista, deriva dal coinvolgimento delle piccole arterie
che forniscono il nervo ottico [97 - 100].Visual
loss occurs in 15-20% of patients [ 97 - 100
]. Perdita della vista si verifica nel 15-20% dei pazienti [97 - 100].In
80-90% of cases visual impairment is due to anterior ischemic optic neuritis
secondary to involvement of the posterior cilliary arteries supplying the optic
nerve [ 101
, 102
]. Nel 80-90% dei casi minorazione visiva è dovuta ad anteriore
ischemica neurite ottica secondaria al coinvolgimento delle arterie posteriori
ciliare che riforniscono il nervo ottico [101,102].Occlusion
of the retinal artery is less frequent and underlies visual loss in 10% of
cases [ 99 , 100
]. Occlusione dell'arteria retinica è meno frequente e sottende
perdita visiva nel 10% dei casi [99,100].
Ischemic stroke or multiinfarct dementia occurs in 3-6% of
patients and is due to inflammatory involvement of the intracranial branches of
the carotid and vertebral arteries. Ictus
ischemico o demenza multiinfarct verifica nel 3-6% dei pazienti ed è dovuto al
coinvolgimento infiammatorio dei rami intracranici della carotide e arterie
vertebrali. [ 97 , 100
, 103
, 104
].[97,100,103,104].When
explored, ultrasonography of the supraaortic branches are frequently normal [ 103
, 104
]. Quando esplorato, ecografia dei rami supraaortic sono spesso
normale [103,104].Usually,
inflammation is limited to the most proximal, extradural part of these arteries.
Solitamente, l'infiammazione è limitata alla più prossimale, parte
extraduraledello queste arterie. In some series,
strokes are more frequent in the vertebrobasilar territories contrarily to
atherosclerotic occlusions which are more frequent in the carotid branches [ 103
]. In alcune serie, i colpi sono più frequenti nei territori
vertebro-basilari contrariamente a occlusioni aterosclerotiche che sono più
frequenti tra i rami della carotide [103].Brain
infarcts are frequently multiple, indicating involvement of various branches,
reduced flow from proximal stenosis, distant embolization of proximal thrombi,
or a combination of these [ 97 , 103
, 104
] (Fig. 5A 5A
). Infarti cerebrali sono spesso multiple, indicando il
coinvolgimento di vari rami, flusso ridotto da stenosi prossimale,
embolizzazione lontano di trombi prossimale, o una combinazione di questi [97,103,104] (Fig. 5A 5A).Although thrombosis is uncommonly seen
in temporal artery biopsies, necropsy studies from patients dying from
GCA-related stroke, frequently disclose thrombosis as a precipitating event [ 100
]. Anche se la trombosi è insolitamente visto in biopsie delle
arterie temporali, studi necroscopico da pazienti che muoiono di ictus GCA
legati, spesso rivelano la trombosi come un evento scatenante [100].Mortality
of GCA-related stroke is about 30% [ 103
, 104
]. La mortalità di ictus GCA relativo è di circa il 30% [103,104].
Multiple infarcts in the cerebral pons, cerebellum, and occipital
lobes in a patient with biopsy-proven giant-cell arteritis who developed ataxia
and cognitive impairment after the initiation of glucocorticoid therapy. B
) Cerebral angiography displaying ... Molteplici
infarti nel ponte cerebrali, cervelletto, e lobi occipitali in un paziente con
biopsia arterite temporale che ha sviluppato atassia e deterioramento cognitivo
dopo l'inizio della terapia con glucocorticoidi. B) L'angiografia
cerebrale visualizzazione ...
Stroke is more frequent among individuals with visual loss
indicating that some individuals may be more prone to develop intracranial
involvement and related complications [ 97 , 98 ]. L'ictus è più frequente tra gli individui con perdita della vista
che indicano che alcuni individui possono essere più inclini a sviluppare il
coinvolgimento intracranica e complicanze correlate [97,98].Several
studies indicate that individuals with prominent extracranial large- vessel
involvement are less prone to develop cranial ischemic complications,
suggesting heterogeneity in patterns of vascular targeting by GCA [ 105
- 107
]. Diversi studi indicano che gli individui con prominente
coinvolgimento nave larga extracranica sono meno inclini a sviluppare
complicanze ischemiche del cranio, suggerendo l'eterogeneità nei modelli di
targeting vascolare da GCA [105 - 107].Several
studies indicate that traditional vascular risk factors are more frequent and
the systemic inflammatory response is weaker in patients with GCA-related
ophthalmic and neurologic ischemic complications, making early diagnosis and
follow up more difficult [ 97 - 100
, 108
]. Diversi studi indicano che i tradizionali fattori di rischio
vascolare sono più frequenti e la risposta infiammatoria sistemica è più debole
nei pazienti con complicanze ischemiche oftalmiche e neurologici GCA correlati,
rendendo la diagnosi precoce e di follow-up più difficile [97 - 100,108].High
dose glucocorticoids usually prevent progression of visual impairment.
Glucocorticoidi ad alte dosi di solito impediscono la progressione della
disabilità visiva. Intravenous methylprednisolone
pulses are usually administered in this setting but there is no proof that this
approach is more effective than the standard daily 60 mg dose. Impulsi
metilprednisolone per via endovenosa sono solitamente somministrati in questa
impostazione ma non vi è alcuna prova che questo approccio è più efficace
rispetto alla dose standard giornaliera di 60 mg. In
10-27% of patients presenting with visual symptoms, vision may continue to
deteriorate during the first 1-2 weeks after the beginning of glucocorticoid
treatment [ 99 ]. Nel 10-27% dei pazienti con sintomi visivi, visione può
continuare a deteriorarsi durante le prime 1-2 settimane dopo l'inizio del
trattamento con glucocorticoidi [99].Antiplatelet
or anticoagulant therapy is usually given in these circumstances with variable
results [ 99 , 101
, 102
]. Antipiastrinica o terapia anticoagulante viene di solito dato in
queste circostanze, con risultati variabili [99,101,102].After
this initial period, the risk of developing subsequent disease-related visual
loss is low, about 1% in 5 years [ 109
]. Dopo questo periodo iniziale, il rischio di sviluppare la
successiva perdita visiva correlata alla malattia è bassa, circa l'1% in 5 anni
[109].
Stroke frequently occurs during the first weeks after the
initiation of glucocorticoid treatment. Ictus si
verifica frequentemente durante le prime settimane dopo l'inizio del
trattamento con glucocorticoidi. Besides adding antiaggregants,
anticoagulants, or both, the classical approach to this situation has been
intensifying glucocorticoid and immunosuppressive therapy. Oltre ad
aggiungere antiaggreganti, anticoagulanti, o entrambi, l'approccio classico di
questa situazione ha intensificato terapia con glucocorticoidi e
immunosoppressiva. However, a recent report indicate
that some patients with proximal lesions may better benefit from intracerebral
percutaneous angioplasty [ 57 ] (Fig 5B 5B
). Tuttavia, un recente rapporto indica che alcuni pazienti con
lesioni prossimali possano meglio beneficiare intracerebrale angioplastica
percutanea [57] (Fig 5B 5B).
4.2.2. 4.2.2. Takayasu Arteritis Arterite di Takayasu
Non specific neurologic manifestations such as headache,
dizziness of variable intensity, and lightheadedness are highly frequent in
patients with Takayasu's arteritis, occurring in 57-90% in most series [ 58 , 110
, 111
] (Fig. 6 6
). Manifestazioni non specifiche neurologiche come mal di testa,
vertigini di intensità variabile, e vertigini sono molto frequenti nei pazienti
con arterite di Takayasu, che si verificano nel 57-90% nella maggior parte
delle serie [58,110,111] (Fig. 6 6).More severe complications include visual disturbances or
visual loss, syncope, transient ischemic attacks and stroke.
Complicazioni più gravi comprendono disturbi visivi o perdita della vista,
sincope, attacchi ischemici transitori e ictus. Most
of these symptoms/complications can be related to extracranial steno-occlusive
lesions in the subclavian (with subsequent arm-steal syndrome), carotid and
vertebral arteries which results in decrease brain flow [ 112
, 113
]. La maggior parte di questi sintomi / complicazioni possono essere
correlati a lesioni steno-occlusiva extracranica in succlavia (con conseguente
sindrome di braccio-rubare), carotide e arterie vertebrali che si traduce in
riduzione del flusso cerebrale [112,113].Stroke
occurs in less than 10% of cases in large cohorts but it is among the leading
causes of premature death in these patients [ 58 ]. Ictus si verifica in meno del 10% dei casi in grandi coorti ma è
tra le principali cause di morte prematura in questi pazienti [58].Strokes
are usually ischemic and secondary thrombosis of stenotic vessels with
subsequent embolization may be precipitating events. Strokes sono di
solito la trombosi ischemico e secondaria dei vasi stenotiche con successiva
embolizzazione può essere eventi precipitanti. It is
important to remark that cardiomyopathy secondary to aortic valve insufficiency
due to aortic root dilatation or hypertension occurs in about 10% of patients
with Takayasu's disease and may also result in thromboembolic strokes [ 58 ]. E 'importante sottolineare che cardiomiopatia secondaria a
insufficienza valvolare aortica a causa di dilatazione della radice aortica o
ipertensione si verifica in circa il 10% dei pazienti con malattia di Takayasu
e può anche portare a ictus tromboembolici [58].Hemorrhagic
stroke related to hypertension has also been reported [ 112
]. Ictus emorragico correlato all'ipertensione è stato segnalato
anche [112].
Multiple stenoses in the carotid and vertebral arteries in a
38-year old patient with Takayasu disease complaining from lightheadedness and
slight dizziness. Stenosi multiple nella
carotide e arterie vertebrali in un paziente di 38 anni con malattia di
Takayasu lamentarsi da stordimento e vertigini. Leggero
Intracranial artery involvement seems to be uncommon. Coinvolgimento dell'arteria intracranica sembra essere raro. A recent prospective study using ultrasonography and MRI
in 17 patients with neurologic symptoms, disclosed signs of intracranial
involvement in 7 patients [ 113
]. Un recente studio prospettico con ecografia e la risonanza
magnetica in 17 pazienti con sintomi neurologici, divulgato segni di
coinvolgimento intracranica in 7 pazienti [113].However,
no angiography was performed and it was not possible to discern whether these
findings were related to vasculitis or previous embolization. Tuttavia,
non l'angiografia è stata effettuata e non era possibile discernere se questi
risultati hanno riguardato vasculite o embolizzazione precedente. Autopsy studies including the brain are scarce in
Takayasu's disease, but intracranial involvement seems to be unusual.
Studi autoptici compreso il cervello sono scarsi nella malattia di Takayasu, ma
il coinvolgimento intracranica sembra essere insolito. However, at least one patient with vasculitis of intracranial arteries
has been reported [ 114
]. Tuttavia, almeno un paziente con vasculite delle arterie
intracraniche stato segnalato [114].
Glucocorticoids and in most instances immunossupressive agents
are mandatory to induce and maintain remission in patients with Takayasu
disease. I glucocorticoidi e nella maggior parte dei casi gli agenti
immunosoppressiva sono obbligatori per indurre e mantenere la remissione nei
pazienti con malattia di Takayasu. Cyclophosphamide
and methotrexate have been useful in open-label studies and mycophenolate has
been also tried in small case series [ 58 , 110
, 115
]. Ciclofosfamide e metotressato sono stati utili in studi in aperto
e micofenolato è stato provato anche in serie caso piccolo [58,110,115].Because
of its side effects cyclophosphamide is usually avoided and other immunosuppressive
agents are preferred, since Takayasu disease is a relapsing condition usually
targeting young women [ 58 , 110
, 115
]. A causa dei suoi effetti collaterali ciclofosfamide è di solito
evitata e di altri agenti immunosoppressivi sono preferiti, dal momento che la
malattia di Takayasu è una condizione recidivante solitamente di mira giovani
donne [58,110,115].TNF
blockade has provided benefit to patients refractory to other therapies [ 116
]. TNF blocco ha fornito beneficio per i pazienti refrattari ad
altre terapie [116].Angioplasty,
stenting and by-pass surgery are very important in the management of severe
neurological involvement [ 56 , 58 ]. Angioplastica, stenting e by-pass chirurgia sono molto importante
nella gestione di grave coinvolgimento neurologico [56,58].For
better results, revascularization procedures, should be avoided, when possible,
during periods of active disease and be performed to patients in remission [ 115
]. Per ottenere risultati migliori, procedure di
rivascolarizzazione, dovrebbe essere evitato, quando possibile, durante i
periodi di malattia attiva ed è realizzato per i pazienti in remissione [115].
CONCLUSIONS CONCLUSIONI
CNS vasculitis, either primary or complicating systemic
vasculitis is uncommon. CNS vasculite, sia primaria o
complicare vasculite sistemica è rara. However CNS
involvement is a major determinant of severity, morbidity and mortality in
patients with vasculitis. Tuttavia il coinvolgimento del sistema nervoso
centrale è un importante determinante di severità, la morbilità e la mortalità
nei pazienti con vasculite. Diagnosis of PACNS is a
challenge and requires high index of clinical suspicion. La diagnosi di
PACNS è una sfida e richiede alto indice di sospetto clinico. Diagnosis is supported by neuroimaging and histologic data
but requires exclusion of other conditions with the appropriate work-up.
La diagnosi è supportata da dati di neuroimaging e istologiche ma richiede
l'esclusione di altre condizioni con l'appropriato work-up. Neuroimaging techniques are pivotal not only to support
the diagnosis but also in the follow up of affected patients. Tecniche
di neuroimaging sono fondamentali non solo per supportare la diagnosi, ma anche
nel follow up dei pazienti affetti. PACNS or CNS
involvement by systemic vasculitis requires prompt recognition and aggressive
treatment in order to reduce mortality and preserve function. PACNS o
CNS coinvolgimento da vasculite sistemica richiede tempestiva identificazione e
trattamento aggressivo al fine di ridurre la mortalità e la funzione di
preservare.
RINGRAZIAMENTI
The authors are grateful to Drs Leonard Calabrese and Carlo
Salvarani for their contributions to the field and for providing illustrative
figures.
Supported by Marató TV3 (MTV3 06/0710) and Ministerio de Ciencia e
Innovación (SAF 08/04328)
RIFERIMENTI
1. Calabrese LH, Mallek JA. Primary
angiitis of the central nervous system, report of 8 new cases: review of the
literature and proposal for diagnostic
criteria. Medicine. 1988; 67 :20–39. [ PubMed ]
2. Birnbaum J, Hellmann DB. Primary
angiitis of the central nervous system. Arch.
Neurol. 2009; 66 :704–709. [ PubMed ]
3. Lie JT. Classification and
histopathologic spectrum of central nervous system vasculitis. Neurol.
Clin.1997; 15 :805–819. [ PubMed ]
4. Sigal LH. The neurologic
presentation of vasculitic and rheumatologic syndromes. A
review. Medicine.1987; 66 :157–180. [ PubMed ]
5. Scolding NJ. Central Nervous System
Vasculitis. Semin.
Immunopathol. 2009; 31 :527–536. [ PubMed ]
6. Benseler SM, Silverman E, Aviv RI,
Schneider R, Armstrong D, Tyrrell PN, deVeber G. Primary vasculitis of the
central nervous system in children. Arthritis.
Rheum. 2006; 54 :1291–1297. [ PubMed ]
7. Elbers J, Benseler SM. Central
nervous system vasculitis in children. Curr. Opin. Rheumatol. 2008; 20:47–54. [ PubMed ]
8. Cantez S, Benseler SM. Childhood
central nervous system vasculitis: a treatable cause of new neurological
deficit in children. Nat. Clin. Pract.
Rheumatol. 2008; 4 :460–461. [ PubMed ]
9. Salvarani C, Brown RD, Jr, Calamia
KT, Christianson TJ, Weigand SD, Miller DV, Giannini C, Meschia JF, Huston J,
3rd, Hunder GG. Primary central nervous system vasculitis, analysis of 101
patients. Ann. Neurol. 2007; 62 :442–451. [ PubMed ]
10. Amlie-Lefond C,
Kleinschmidt-DeMasters BK, Mahalingam R, Davis LE, Gilden DH. The vasculopathy
of varicella-zoster virus encephalitis. Ann. Neurol. 1995; 37 :784–790. [ PubMed ]
11. Yankner BA, Skolnik PR, Shoukimas
GM, Gabuzda DH, Sobel RA, Ho DD. Cerebral granulomatous angiitis associated
with isolation of human T-lymphotropic virus type III from the central nervous
system.Ann. Neurol. 1986; 20 :362–364. [ PubMed ]
12. Gray F, Hurtrel M, Hurtrel B.
Early central nervous system changes in human immunodeficiency virus
(HIV)-infection. Neuropathol. Appl. Neurobiol. 1993; 19 :3–9. [ PubMed ]
13. Reuter JD. Cytomegalovirus induces
T-cell independent apoptosis in brain during
immunodeficiency. J.Clin. Virol. 2005; 32 :218–223. [ PubMed ]
14. Linnemann CC, Jr, Alvira M.
Pathogenesis of varicella-zoster angiitis in the CNS. Arch.
Neurol. 1980;37 :239–240. [ PubMed ]
15. Calabrese LH. Infection with the
human immunodeficiency virus type 1 and vascular inflammatory
disease. Clin. Exp. Rheumatol. 2004; 22 (suppl):S87–93. [ PubMed ]
16. Lie JT. Primary (granulomatous)
angiitis of the central nervous system: a clinicopathologic analysis of 15 new
cases and a review of the literature. Hum.
Pathol. 1992; 23 :164–171. [ PubMed ]
17. Sethna MP, Lampson LA. Immune
modulation within the brain, recruitment of inflammatory cells and increased
major histocompatibility antigen expression following intracerebral injection
of interferon-gamma.J.
Neuroimmunol. 1991; 34 :121–32. [ PubMed ]
18. Akassoglou K, Douni E, Bauer J,
Lassmann H, Kollias G, Probert L. Exclusive tumor necrosis factor (TNF)
signaling by the p75TNF receptor triggers inflammatory ischemia in the CNS of
transgenic
mice.Proc. Natl. Acad. Sci. USA. 2003; 100 :709–714. [ PMC free article ] [ PubMed ]
19. Hirohata S, Tanimoto K, Ito K.
Elevation of cerebrospinal fluid interleukin-6 activity in patients with
vasculitides and central nervous system involvement. Clin. Immunol.
Immunopathol. 1993; 66 :225–229.[ PubMed ]
20. Miller DV, Salvarani C, Hunder GG,
Brown RD, Parisi JE, Christianson TJ, Giannini C. Biopsy findings in primary
angiitis of the central nervous system. Am. J. Surg.
Pathol. 2009; 33 :35–43. [ PubMed ]
21. Myung J, Kim B, Yoon BW, Lee SK,
Sung JJ, Chung CK, Chang KH, Park SH. B-cell dominant lymphocytic primary
angiitis of the central nervous system, Four biopsy-proven
cases. Neuropathology.2010; 30 :123–130. [ PubMed ]
22. Calabrese LH, Duna GF, Lie JT.
Vasculitis in the central nervous system. Arthritis
Rheum. 1997; 40:1189–1201. [ PubMed ]
23. Scolding NJ, Jayne DR, Zajicek JP,
Meyer PAR, Wraight EP, Lockwood CM. The syndrome of cerebral vasculitis,
recognition; diagnosis and management. QJ
Med. 1997; 90 :61–73. [ PubMed ]
24. MacLaren K, Gillespie J, Shrestha
S, Neary D, Ballardie FW. Primary angiitis of the central nervous system:
emerging variants. QJ Med. 2005; 98 :643–654. [ PubMed ]
25. Molloy ES, Singhal AB, Calabrese
LH. Tumour-like mass lesion, an under-recognised presentation of primary
angiitis of the central nervous system. Ann. Rheum.
Dis. 2008; 67 :1732–1735. [ PubMed ]
26. Salvarani C, Brown RD, Calamia KT,
Christianson TJ, Huston J 3rd, Meschia JF, Giannini C, Miller DV, Hunder GG.
Primary CNS vasculitis with prominent leptomeningeal enhancement. A subset with
a benign outcome. Arthritis. Rheum. 2008; 58 :595–603. [ PubMed ]
27. Salvarani C, Brown RD, Calamia KT,
Christianson TJ, Huston J 3rd, Meschia JF, Giannini C, Miller DV, Hunder GG.
Angiography negative primary central nervous system vasculitis. A syndrome
involving small cerebral vessels. Medicine. 2008; 87 :264–271. [ PubMed ]
28. Hajj-Ali RA, Calabrese L. Central
nervous system vasculitis. Curr. Opin.
Rheumatol. 2009; 21 :10–18.[ PubMed ]
32. Hellmann DB, Roubenoff R, Healy
RA, Wang H. Central nervous system angiography, Safety and predictors of a
positive result in 125 consecutive patients evaluated for possible
vasculitis. J. Rheumatol.1992; 19 :568–572. [ PubMed ]
33. Cloft HJ, Phillips CD, Dix JE,
McNulty BC, Zagardo MT, Kallmes DF. Correlation of angiography and MR imaging
in cerebral vasculitis. Acta.
Radiol. 1999; 40 :83–87. [ PubMed ]
34. Stone JH, Pomper MG, Roubenoff R,
Mille TJ, Hellmann DB. Sensitivities of noninvasive tests for central nervous
system vasculitis, a comparison of lumbar puncture; computed tomography; and
magnetic resonance imaging. J.
Rheumatol. 1994; 21 :1277–1282. [ PubMed ]
35. White ML, Hadley WL, Zhang Y,
Dogar MA. Analysis of central nervous system vasculitis with diffusion-weighted
imaging and apparent diffusion coefficient mapping of the normal-appearing
brain. Am. J. Neuroradiol. 2007; 28 :933–937. [ PubMed ]
36. Kraemer M, Berlit P. Primary
central nervous system vasculitis: clinical experiences with 21 new European
cases. Rheumatol. Int. 2010; 257 (7):816–9. [ PubMed ]
37. Kraemer M, Berlit P. Systemic,
secondary and infectious causes for cerebral vasculitis: clinical experience
with 16 new European cases. Rheumatol.
Int. 2010; 30 :1471–1476. [ PubMed ]
38. Vassallo R, Remstein E, Parisi JE,
Huston J, 3rd, Brown RD., Jr Multiple cerebral infarctions from nonbacterial
thrombotic endocarditis mimicking cerebral vasculitis. Mayo Clin.
Proc. 1999; 74 :798–802.[ PubMed ]
39. Salvarani C, Brown RD, Jr, Calamia
KT, Christianson TJ, Huston J, 3rd, Meschia JF, Giannini C, Miller DV, Hunder
GG. Primary central nervous system vasculitis: comparison of patients with and
without cerebral amyloid
angiopathy. Rheumatology. 2008; 47 :1671–1677. [ PubMed ]
40. Mukhtyar C, Guillevin L, Cid MC,
Dasgupta B, de Groot K, Gross W, Hauser T, Hellmich B, Jayne D, Kallenberg CG,
Merkel PA, Raspe H, Salvarani C, Scott DG, Stegeman C, Watts R, Westman K,
Witter J, Yazici H, Luqmani R. European Vasculitis Study Group. EULAR
recommendations for the management of primary medium vessel
vasculitis. Ann. Rheum. Dis. 2009; 68 :310–317. [ PubMed ]
41. de Groot K, Harper L, Jayne DR,
Flores Suarez LF, Gregorini G, Gross WL, Luqmani R, Pusey CD, Rasmussen N,
Sinico RA, Tesar V, Vanhille P, Westman K, Savage CO. EUVAS (European
Vasculitis Study Group) Pulse versus daily oral cyclophosphamide for induction
of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a
randomized
trial. Ann. Intern. Med. 2009; 150 :670–80.[ PubMed ]
42. Jayne D, Rasmussen N, Andrassy K,
Bacon P, Tervaert JW, Dadoniené J, Ekstrand A, Gaskin G, Gregorini G, de Groot
K, Gross W, Hagen EC, Mirapeix E, Pettersson E, Siegert C, Sinico A, Tesar V,
Westman K, Pusey C. European Vasculitis Study Group. A randomized trial of
maintenance therapy for vasculitis associated with antineutrophil cytoplasmic
autoantibodies. N. Engl. J.
Med. 2003; 349 :36–44.[ PubMed ]
43. Pagnoux C, Mahr A, Hamidou MA,
Boffa JJ, Ruivard M, Ducroix JP, Kyndt X, Lifermann F, Papo T, Lambert M, Le
Noach J, Khellaf M, Merrien D, Puéchal X, Vinzio S, Cohen P, Mouthon L, Cordier
JF, Guillevin L. French Vasculitis Study Group. Azathioprine or methotrexate
maintenance for ANCA-associated vasculitis. N. Engl. J.
Med. 2008; 359 :2790–2803. [ PubMed ]
44. Jones RB, Cohen-Tervaert JW,
Hauser T, Luqmani R, Morgan MD, Peh CA, Savage CO, Segelmark M, Tesar V, van
Paassen P, Walsh D, Walsh M, Westman K, Jayne DR. European Vasculitis Study
Group. Rituximab versus cyclophosphamide in ANCA-associated renal
vasculitis. N. Engl. J.
Med. 2010; 363 :211–220. [ PubMed ]
45. Stone JH, Merkel PA, Spiera R, Seo
P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK,
Webber L, Ding L, Sejismundo LP, Mieras K, Weitzenkamp D, Ikle D,
Seyfert-Margolis V, Mueller M, Brunetta P, Allen NB, Fervenza FC, Geetha D,
Keogh KA, Kissin EY, Monach PA, Peikert T, Stegeman C, Ytterberg SR, Specks U.
RAVE-ITN Research Group. Rituximab versus cyclophophamide for ANCA-associated
vasculitis. N. Eng. J. Med. 2010; 363 :221–232. [ PMC free article ][ PubMed ]
46. Tokunaga M, Saito K, Kawabata D,
Imura Y, Fujii T, Nakayamada S, Tsujimura S, Nawata M, Iwata S, Azuma T, Mimori
T, Tanaka Y. Efficacy of rituximab (anti-CD20) for refractory systemic lupus
erythematosus involving the central nervous system. Ann. Rheum.
Dis. 2007; 66 :470–475.[ PMC free article ] [ PubMed ]
47. Salvarani C, Brown RD, Jr, Calamia
KT, Huston J, 3rd, Meschia JF, Giannini C, Miller DV, Hunder GG. Efficacy of
tumor necrosis factor alpha blockade in primary central nervous system
vasculitis resistant to immunosuppressive treatment. Arthritis
Rheum. 2008; 59 :291–296. [ PubMed ]
49. Ducros A, Boukobza M, Porcher R,
Sarov M, Valade D, Bousser MG. The clinical and radiological spectrum of
reversible cerebral vasoconstriction syndrome. A prospective series of 67
patients. Brain. 2007;130 :3091–3101. [ PubMed ]
50. Hajj-Ali RA, Furlan A, Abou-Chebel
A, Calabrese LH. Benign angiopathy of the central nervous system, cohort of 16
patients with clinical course and long-term followup. Arthritis.
Rheum. 2002; 47 :662–669. [ PubMed ]
51. Lu SR, Liao YC, Fuh JL, Liang JF,
Wang SJ. Nimodipine for treatment of primary thunderclap
headache. Neurology. 2004; 62 :414–1416. [ PubMed ]
52. Calabrese LH, Molloy ES, Singhal
AB. Primary central nervous system vasculitis: progress and
questions. Ann. Neurol. 2007; 62 :430–432. [ PubMed ]
53. Zivkovic S, Moore PM. Systemic and
central nervous system vasculitis. Curr. Treat. Options
Neurol.2000; 2 :459–472. [ PubMed ]
54. Guillevin L, Lhote F, Gayraud M,
Cohen P, Jarrousse B, Lortholary O, Thibult N, Casassus P. Prognostic factors
in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342
patients.Medicine (Baltimore) 1996; 75 :17–28. [ PubMed ]
55. Bourgarit A, Le Toumelin P, Pagnoux
, Cohen P, Mahr A, Le Guern V, Mouthon L, Guillevin L. French Vasculitis Study
Group. Deaths occurring during the first year after treatment onset for
polyarteritis nodosa; microscopic polyangiitis; and Churg- Strauss syndrome, a
retrospective analysis of causes and factors predictive of mortality based on
595 patients. Medicine
(Baltimore) 2005; 84 :323–330. [ PubMed ]
56. Mukhtyar C, Guillevin L, Cid MC,
Dasgupta B, de Groot K, Gross W, Hauser T, Hellmich B, Jayne D, Kallenberg CG,
Merkel PA, Raspe H, Salvarani C, Scott DG, Stegeman C, Watts R, Westman K,
Witter J, Yazici H, Luqmani R. European Vasculitis Study Group. EULAR
recommendations for the management of large- vessel vasculitis. Ann.
Rheum. Dis. 2009; 68 :318–323. [ PubMed ]
57. Espígol-Frigolé G, Gómez-Choco M,
Obach V, Sanroman L, Prieto S, Argelich R, Grau JM, Cid MC. Percutaneous
transluminal angioplasty of internal carotid and vertebral arteries in
giant-cell arteritis: report of 2
cases. APMIS. 2009; 117 (suppl):104–105.
58. Maksimowicz-McKinnon K, Clark TM,
Hoffman GS. Limitations of therapy and a guarded prognosis in an American
cohort of Takayasu arteritis patients. Arthritis
Rheum. 2007; 56 :1000–1009. [ PubMed ]
59. Hoffman GS, Kerr GS, Leavitt RY,
Hallahan CW, Lebovics RS, Travis WD, Rottem M, Fauci AS. Wegener
granulomatosis: an analysis of 158
patients. Ann. Intern. Med. 1992; 116 :488–498. [ PubMed ]
60. Reinhold-Keller E, Beuge N, Latza
U, de Groot K, Rudert H, Nölle B, Heller M, Gross WL. An interdisciplinary
approach to the care of patients with Wegener's granulomatosis, long-term
outcome in 155 patients. Arthritis Rheum. 2000; 43 :1021–1032. [ PubMed ]
61. Stone JH. Wegener's Granulomatosis
Etanercept Trial Research Group. Limited versus severe Wegener's
granulomatosis, baseline data on patients in the Wegener's granulomatosis
etanercept trial. Arthritis
Rheum.2003; 48 :2299–2309. [ PubMed ]
62. Nishino H, Rubino FA, DeRemee RA,
Swanson JW, Parisi JE. Neurological involvement in Wegener's granulomatosis: an
analysis of 324 consecutive patients at the Mayo Clinic. Ann.
Neurol. 1993; 33 :4–9.[ PubMed ]
63. Seror R, Mahr A, Ramanoelina J,
Pagnoux C, Cohen P, Guillevin L. Central nervous system involvement in Wegener
granulomatosis. Medicine (Baltimore) 2006; 85 :54–65. [ PubMed ]
64. Di Comite G, Bozzolo EP, Praderio L, Tresoldi M,
Sabbadini MG. Meningeal involvement in
Wegener's granulomatosis is associated with localized disease. Clin. Exp. Rheumatol. 2006; 24 :S60–64. [ PubMed ]
65. Di Comite G, Bozzolo E, Bianchi S, Sabbadini MG. Two cases of meningeal involvement in Wegener's
granulomatosis. Rheumatology
(Oxford) 2004; 43 :1459–1460. [ PubMed ]
66. Waqqas S. Case records of the
Massachusetts General Hospital. Weekly clinicopathological exercises. Case
9-1999. A 74-year-old woman with hydrocephalus and
pleocytosis. N. Engl. J. Med. 1999; 340 :945–953. [ PubMed ]
67. Guillevin L, Durand-Gasselin B,
Cevallos R, Gayraud M, Lhote F, Callard P, Amouroux J, Casassus P, Jarrousse B.
Microscopic polyangiitis, clinical and laboratory findings in eighty-five
patients. Arthritis Rheum. 1999; 42 :421–430. [ PubMed ]
69. Sasaki A, Hirato J, Nakazato Y,
Tanaka T, Takeuchi H. An autopsy case of P-ANCA-positive microscopic
polyangiitis with multiple cerebral hemorrhagic infarction. No To
Shinkei. 1998; 50 :56–60.[ PubMed ]
70. Tang CW, Wang PN, Lin KP, Huang
DF, Wang SJ, Chen WT. Microscopic polyangiitis presenting with capsular warning
syndrome and subsequent
stroke. J. Neurol. Sci. 2009; 277 :174–175. [ PubMed ]
71. Furukawa Y, Matsumoto Y, Yamada M.
Hypertrophic pachymeningitis as an initial and cardinal manifestation of
microscopic
polyangiitis. Neurology. 2004; 63 :1722–1724. [ PubMed ]
72. Kono H, Inokuma S, Nakayama H,
Yamazaki J. Pachymeningitis in microscopic polyangiitis (MPA), a case report
and a review of central nervous system involvement in
MPA. Clin. Exp. Rheumatol. 2000; 18:397–400. [ PubMed ]
73. Keogh KA, Specks U. Churg-Strauss
syndrome, clinical presentation; antineutrophil cytoplasmic antibodies; and
leukotriene receptor antagonists. Am. J.
Med. 2003; 115 :284–290. [ PubMed ]
74. Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A,
Tosoni C, Grasselli C, Pavone L, Gregorini G, Monti S, Frassi M, Vecchio F,
Corace C, Venegoni E, Buzio C. Prevalence and clinical significance of
antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 2005; 52 :2926–2935.[ PubMed ]
75. Guillevin L, Cohen P, Gayraud M,
Lhote F, Jarrousse B, Cassasus P. Churg-Strauss syndrome, clinical study and
long-term follow-up of 96
patients. Medicine. 1999; 78 :26–37. [ PubMed ]
76. Sablé-Fourtassou R, Cohen P, Mahr
A, Pagnoux C, Mouthon L, Jayne D, Blockmans D, Cordier JF, Delaval P, Puechal
X, Lauque D, Viallard JF, Zoulim A, Guillevin L. French Vasculitis Study Group.
Antineutrophil cytoplasmic antibodies and the Churg-Strauss
syndrome. Ann. Intern. Med. 2005; 143 :632–638. [ PubMed ]
77. Solans R, Bosch JA,
Pérez-Bocanegra C, Selva A, Huguet P, Alijotas J, Orriols R, Armadans L,
Vilardell M. Churg-Strauss syndrome, outcome and long-term follow-up of 32
patients. Rheumatology.2001; 40 :763–771. [ PubMed ]
79. Sheerin UM, Barreto J, Brown MM,
Brew S, Losseff NA. Subarachnoid haemorrhage as the first clinicalmanifestation
of Churg-Strauss syndrome. J.
Neurol. 2008; 255 :607–608. [ PubMed ]
80. Tokumaru AM, Obata T, Kohyama S,
Kaji T, Okizuka H, Suzuki K, Kusano S. Intracranial meningeal involvement in
Churg-Strauss syndrome. Am. J.
Neuroradiol. 2002; 23 :221–224. [ PubMed ]
81. Lio M, Fukuda S, Maguchi S,
Kawanami M, Inuyama Y. Churg-Strauss syndrome with pachymeningitis refractory
to steroid therapy alone-a case report. Auris Nasus
Larynx. 2001; 28 :S121–125. [ PubMed ]
82. Pagnoux C, Seror R, Henegar C,
Mahr A, Cohen P, Le Guern V, Bienvenu B, Mouthon L, Guillevin L. French Vasculitis
Study Group. Clinical features and outcomes in 348 patients with polyarteritis
nodosa, a systematic retrospective study of patients diagnosed between 1963 and
2005 and entered into the French Vasculitis Study Group
Database. Arthritis Rheum. 2010; 62 :616–626. [ PubMed ]
83. Rosenberg MR, Parshley M, Gibson
S, Wernick R. Central nervous system polyarteritis nodosa. West. J.
Med. 1990; 153 :553–556. [ PMC free article ] [ PubMed ]
84. Basu N, Watts R, Bajema I, Baslund
B, Bley T, Boers M, Brogan P, Calabrese L, Cid MC, Cohen-Tervaert JW,
Flores-Suarez LF, Fujimoto S, de Groot K, Guillevin L, Hatemi G, Hauser T,
Jayne D, Jennette C, Kallenberg CG, Kobayashi S, Little MA, Mahr A, McLaren J,
Merkel PA, Ozen S, Puechal X, Rasmussen N, Salama A, Salvarani C, Savage C,
Scott DG, Segelmark M, Specks U, Sunderköetter C, Suzuki K, Tesar V, Wiik A,
Yazici H, Luqmani R. EULAR points to consider in the development of
classification and diagnostic criteria in systemic vasculitis. Ann. Rheum.
Dis. 2010; 69 :1744–1750.[ PubMed ]
85. Provenzale JM, Allen NB.
Neuroradiologic findings in polyarteritis nodosa. Am. J.
Neuroradiol. 1996;17 :1119–1126. [ PubMed ]
86. Case records of the Massachusetts
General Hospital. Weekly clinicopathological exercises. Case 3-2003. A
36-year-old man with renal failure; hypertension and neurologic
abnormalities. N. Engl. J.
Med. 2003; 348:333–342. [ PubMed ]
87. Ramos-Casals M, Robles A,
Brito-Zerón P, Nardi N, Nicolas JM, Forns X, Plaza J, Yagüe J, Sánchez-Tapias
JM, Font J. Life- threatening cryoglobulinemia: clinical and immunologic
characterization of 29 cases.Semin. Artritis
Rheum. 2006; 36 :189–196. [ PubMed ]
88. Casato M, Saadoun D, Marchetti A,
Limal N, Picq C, Pantano P, Galanaud D, Cianci R, Duhaut P, Piette JC, Fiorilli
M, Cacoub P. Central nervous system involvement in hepatitis C virus
cryoglobulinemia vasculitis, a multicenter case-control study using magnetic
resonance imaging and neuropsychological tests.J.
Rheumatol. 2005; 32 :484–488. [ PubMed ]
89. Origgi L, Vanoli M, Carbone A,
Grasso M, Scorza R. Central nervous system involvement in patients with
HCV-related cryoglobulinemia. Am. J.
Med. Sci. 1998; 315 :208–210. [ PubMed ]
90. Petty GW, Duffy J, Huston J.
Cerebral ischemia in patients with hepatitis C virus infection and mixed
cryoglobulinemia. Mayo Clin.
Proc. 1996; 71 :671–678. [ PubMed ]
91. Al-Araji A, Sharquie K, Al-Rawi Z.
Prevalence and patterns of neurological involvement in Behçet's disease, a
prospective study from Iraq. J. Neurol. Neurosurg.
Psychiatry. 2003; 74 :608–613.[ PMC free article ] [ PubMed ]
92. Ashjazadeh N, Borhani Haghighi A,
Samangooie S, Moosavi H. Neuro-Behçet's disease, a masquerader of multiple
sclerosis. A prospective study of neurologic manifestations of Behçet's disease
in 96 Iranian
patients. Exp. Mol. Pathol. 2003; 74 :17–22. [ PubMed ]
93. Al-Araji A, Kidd DP.
Neuro-Behçet's disease, epidemiology; clinical characteristics; and
management.Lancet Neurol. 2009; 8 (2):192–204. [ PubMed ]
94. Akman-Demir G, Serdaroglu P, Tasçi
B. Clinical patterns of neurological involvement in Behçet's disease,
evaluation of 200 patients. The Neuro-Behçet Study
Group. Brain. 1999; 122 :2171–2182.[ PubMed ]
95. Lee SH, Yoon PH, Park SJ, Kim DI.
MRI findings in neuroBehçet's disease. Clin.
Radiol. 2001; 56:485–494. [ PubMed ]
96. Esteban MJ, Font C,
Hernández-Rodríguez J, Valls-Solé J, Sanmartí R, Cardellach F, García-Martínez
A, Campo E, Urbano-Márquez A, Grau JM, Cid MC. Small-vessel vasculitis
surrounding a spared temporal artery: clinical and pathological findings in a
series of twenty-eight patients. Arthritis
Rheum. 2001; 44:1387–1395. [ PubMed ]
97. Cid MC, Font C, Oristrell J, de la Sierra A, Coll-Vinent
B, López-Soto A, Vilaseca J, Urbano-Márquez A, Grau JM. Association between strong inflammatory response and low risk of
developing visual loss and other cranial ischemic complications in giant cell
(temporal) arteritis. Arthritis
Rheum. 1998; 41 :26–32.[ PubMed ]
98. González-Gay MA, García-Porrua C,
Llorca J, Hajeer AH, Brañas F, Dababneh A, González-Louzao C, Rodriguez-Gil E,
Rodríguez-Ledo P, Ollier WE. Visual manifestations of giant cell arteritis:
trends and clinical spectrum in 161 patients. Medicine
(Baltimore) 2000; 79 :283–292. [ PubMed ]
99. Cid MC, García-Martínez A, Lozano
E, Espígol-Frigolé G, Hernández-Rodríguez J. Five clinical conundrums in the
management of giant-cell arteritis. Rheum. Dis. Clin. North
Am. 2007; 33 :819–834.[ PubMed ]
100. Cid MC, Prieto-González S, Arguis
P, Espígol-Frigolé G, Butjosa M, Hernández-Rodríguez J, Segarra M, Lozano E,
García-Martínez A. The spectrum of vascular involvement in giant-cell
arteritis: clinical consequences of detrimental vascular remodelling at
different sites. APMIS. 2009; 117 (suppl):10–20.[ PubMed ]
101. Hayreh SS, Zimmerman B. Visual
deterioration in giant cell arteritis patients while on high doses of
glucocorticoid
therapy. Ophthalmology. 2003; 110 :1204–1215. [ PubMed ]
102. Hayreh SS, Zimmerman B, Kardon
RH. Visual improvement with corticosteroid therapy in giant cell arteritis.
Report of a large study and review of literature. Acta Ophthalmol.
Scand. 2002; 80 :353–367.[ PubMed ]
103. Gonzalez-Gay MA,
Vazquez-Rodriguez TR, Gomez-Acebo I, Pego-Reigosa R, Lopez-Diaz MJ,
Vázquez-Triñanes MC, Miranda-Filloy JA, Blanco R, Dierssen T, Gonzalez-Juanatey
C, Llorca J. Strokes at time of disease diagnosis in a series of 287 patients
with biopsy-proven giant cell arteritis. Medicine
(Baltimore) 2009; 88 :227–235. [ PubMed ]
104. Solans-Laqué R, Bosch-Gil JA,
Molina-Catenario CA, Ortega-Aznar A, Alvarez-Sabin J, Vilardell-Tarres M.
Stroke and multi-infarct dementia as presenting symptoms of giant cell
arteritis, report of 7 cases and review of the literature. Medicine
(Baltimore) 2008; 87 :335–344. [ PubMed ]
105. Brack A, Martinez-Taboada V,
Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel
giant cell arteritis. Arthritis
Rheum. 1999; 42 :311–317. [ PubMed ]
106. Nuenninghoff DM, Hunder GG,
Christianson TJ, McClelland RL, Matteson EL. Incidence and predictors of
large-artery complication (aortic aneurysm, aortic dissection, and/or
large-artery stenosis) in patients with giant cell arteritis: a
population-based study over 50 years. Arthritis
Rheum. 2003; 48 :3522–3531. [ PubMed ]
107. Schmidt WA, Seifert A,
Gromnica-Ihle E, Krause A, Natusch A. Ultrasound of proximal upper extremity
arteries to increase the diagnostic yield in large-vessel giant cell
arteritis. Rheumatology. 2008; 47:96–101. [ PubMed ]
108. Salvarani C, Cimino L, Macchioni
P, Consonni D, Cantini F, Bajocchi G, Pipitone N, Catanoso MG, Boiardi L. Risk
factors for visual loss in an Italian population-based cohort of patients with
giant cell arteritis.Arthritis Rheum. 2005; 53 :293–297. [ PubMed ]
110. Kerr GS, Hallahan CW, Giordano J,
Leavitt RY, Fauci AS, Rottem M, Hoffman GS. Takayasu
arteritis. Ann. Intern. Med. 1994; 120 :919–929. [ PubMed ]
111. Vanoli M, Daina E, Salvarani C, Sabbadini MG, Rossi C,
Bacchiani G, Schieppati A, Baldissera E, Bertolini G. Itaka Study Group. Takayasu's arteritis, A study of 104 Italian
patients. Arthritis Rheum. 2005;53 :100–107. [ PubMed ]
112. Kim HJ, Suh DC, Kim JK, Kim SJ,
Lee JH, Choi CG, Yoo B, Kwon SU, Kim JS. Correlation of neurological
manifestations of Takayasu's arteritis with cerebral angiographic
findings. Clin. Imaging. 2005;29 :79–85. [ PubMed ]
113. Ringleb PA, Strittmatter EI,
Loewer M, Hartmann M, Fiebach JB, Lichy C, Weber R, Jacobi C, Amendt K,
Schwaninger M. Cerebrovascular manifestations of Takayasu arteritis in
Europe. Rheumatology.2005; 44 :1012–1015. [ PubMed ]
114. Molnár P, Hegedüs K. Direct
involvement of intracerebral arteries in Takayasu's arteritis. Acta.
Neuropathol. 1984; 63 :83–86. [ PubMed ]
115. Alba MA, Espígol-Frigolé G,
Butjosa M, Prieto-González S, García-Martínez A, Hernández-Rodríguez J, Cid MC.
Treatment of large-vessel vasculitis. Curr. Immunol.
Rev. 2011 (in press)
116. Molloy ES, Langford CA, Clark M,
Gota CE, Hoffman GS. Anti-tumour necrosis factor therapy in patients with
refractory Takayasu arteritis: long-term follow-up. Ann. Rheum. Dis. 2008; 67 :1567–1569.[ PubMed ]
Mentre la maggior parte dei pazienti
che si presentano per i servizi di emergenza (ED) con vertigini isolate hanno
disturbi benigni, circa il 0,7-3% ha un infarto cerebellare(Seemungalbm.2007;. Kerber et al.,2006) perché i sintomi dello infarto
cerebellare si sovrappongono sostanzialmente con le condizioni benigne
l’infarto cerebellare è
comunemente trascurato, con un tasso di diagnosi errate stimata al 35% 2. I pazienti con infarto
cerebellare non diagnosticato in generale sono a più alto rischio di
complicanze, con un tasso di mortalità forse pari al 40% (Savitz et al.,2007).
La diagnosi
fisica è la modalità diagnostica più importante per infarto
cerebellare. Il ricorso a tomografia computerizzata (CT) non è
sufficiente, perché è solo il 26% è sensibile per l’ictus acuto. 4, invece, importanti segni fisici
sono presenti nella maggior parte dei pazienti con infarto Infarto
cerebellare è un sottotipo relativamente rara di ictus ischemico. L’Occlusione di una qualsiasi
delle tre arterie cerebellari che alimentano il cervelletto può determinare un infarto
cerebellare senza interessamento del tronco-encefalo
Tre sono
le maggiori arterie del cervelletto: l’arteria cerebellare postero-inferiore
(PICA), l’arteria cerebellare antero-inferiore (AICA) e l’arteria cerebellar
superiore (SCA). Oltre a permettere il regolare flusso ematico ai distretti del
tronco encefalico, esse irrorano parte del cervelletto. Caratteristica delle
arterie cerebellari è la loro ricca rete di anastomosi per cui l’eventuale
occlusione di una di esse può provocare soltanto una lesione infartuale
limitata alle strutture nervose del tronco encefalico mentre rimane inalterato
il circolo a livello cerebellare. La PICA con i suoi due rami mediale e
laterale irrora il distretto mediale del cervelletto compreso il verme. L’AICA
irrora invece la parte antero-inferiore del cervelletto compreso il flocculo e
il paraflocculo che, com’è noto, hanno un ruolo importante nell’integrazione
delle afferenze visive e vestibolari (Fig. 3B-C).
Ci sono
tre principali arterie che forniscono il cervelletto L SCA (arteria
cerebellare superiore),
Infarti
cerebellari conto di circa il ~ 2% (range 1,5-2,3%) di tutti infarto
cerebrale 1-2.
La
presentazione clinica
Il
quadro clinico di un infarto cerebellare, si manifesta con vertigine acuta,
assai intensa, associata a nausea e vomito sintomi che, inizialmente, possono
orientare verso una patologia improvvisa della periferia vestibolare. Ma la
presenza di segni di interessamento cerebellare: asinergia, dismetria,
ipotonia, disturbi della coordinazione dei movimenti delle estremità, andamento
atassico e di un nistagmo da “direzione dello sguardo” verso un lato e verso il
basso permetteranno di porre una corretta diagnosi. In alcuni pazienti, dopo un
periodo di tempo che varia da i a 4 giorni dalla comparsa della sintomatologia
neurologica, il quadro semeiologico-clinico può progressivamente peggiorare a
causa della compressione esercitata sul tronco encefalico dall’aumento di
volume del cervelletto. Ciò avviene più comunemente nelle lesioni infarmali del
distretto di distribuzione della branca mediale dell’arteria cerebellare
postero-inferiore.
Tab. I - Sintomi e strutture nervose interessate nelle lesioni infartuali nel territorio irrorato dalla P1CA e
dall’AICA
Sintomi
Infarto
nel
territorio
della PICA
Infarto
nel
territorio
della AICA
Vertigine
Nn. vest.,
distretto cereb. post-inf.
Distretto
dorsolat. b-pontino, flocculo, labirinto
Ipoacusia,
acuf.,
vertigine
Assenti
Ipoacusia,
acuf., vertigine
Segni
di atassia -
Presenti
(tratto cereb. ventrale)
Presenti
(ped .cereb. medio)
Disfagia
I
X
Assente
Disfonia
X
Emianest.
facciale omolat -
V
V
Paralisi
del
Presente
Presente
Emi-ipoestesia
controlat.
Presente
(tratto spino-tal.)
Presente
(tratto spino-tal.)
Caratteristiche
radiografiche
CT
Caratteristiche
tipiche di infarto, come la perdita precoce di grigio-bianco differenziazione,
hypoattenuation ed edema, progressione di encefalomalacia cronica possono
essere identificati nei rispettivi territori vascolari.
Il
trattamento delle alterazioni del circolo vertebro-basilare conclamate nella
sindrome ischemica laterale del bulbo o nella sindrome da ischemia cerebellare
sono quelle classiche dell’ictus ischemico acuto. In fase acuta è consigliabile
una TAC cerebrale senza contrasto per distinguere un ictus ischemico da un
ictus emorragico. I trattamenti dell’ictus ischemico si possono raggruppare in
5 categorie: 1) supporto medico; 2) trombolisi; 3) anticoagulanti; 4)
antiaggreganti; 5) neuroprotezione.
Trattamento
e la prognosi
La
mortalità correlata ad infarti cerebellari è superiore a quella di altri
territori vascolari. Questo è generalmente dovuto alla concomitante infarto del
tronco encefalico, o idrocefalo compressione, piuttosto che infarto cerebellare
in sé. Complicanze insoliti includono:
Goal
del trattamento in acuto dell’ictus è quello di ottimizzare la perfusione
cerebrale nella zona ischemia. E essenziale una adeguata assistenza per
prevenire la comparsa di complicanze legate all’evento vasale (trombosi venosa
profonda con embolia polmonare, polmoniti. infezioni urinarie, ulcere da
decubito). La pressione arteriosa non deve essere abbassata drasticamente per
evitare fenomeni di iperperfusione.
La trombolisi
Non
è indicata negli infatti cerebrali acuti del circolo posteriore. In pazienti
selezionati il trattamento trombolitico intra-arterioso della occlusione
dell’arteria basilare potrebbe essere utile, ma la terapia trombolitica
intra-arteriosa è attualmente solo sperimentale.
Terapia con anticoagulanti
L’uso
di anticoagulanti (eparina in bolo) nel trattamento della ischemia cerebrale
aterotrombotica è controverso. L’eparina è però utilizzata in caso di
dissezione senza evidenza clinica supportata da trials clinici controllati.
Terapia con antiaggreganti
L’ictus
in fase acuta va trattato elettivamente con antiaggreganti. L’aspirina rimane
il farmaco di prima scelta alla dose compresa fra 100-300 mg al giorno.
L’effetto dell’aspirina è immediato (circa un’ora dopo l’assunzione) e dura il
tempo di vita delle piastrine (7-10 giorni).
In alternativa all’aspirina, in pazienti che abbiano avuto altri episodi di
ischemia acuta transitoria
in corso
di trattamento con aspirina. o in caso di intulleranza si possono prendere in
considerazione altri antiaggreganti piastrinici come la ticlopidina (250 mg per
2 volte al dì) ed il clopidogrel (75 mg al giorno).
Neuroprotezione 11 concetto di neuroprotezione implica tutte quelle misure atte ad
aumentare la soglia di tolleranza del tessuto cerebrale all’insulto ischemico.
L’unica misura che in un contesto sperimentale abbia dimostrato attualmente
qualche utilità è l’ipotermia controllata che non viene utilizzata in un
contesto clinico.
Molti
dei sintomi di infarto cerebellare sono non-specifici, come nausea, vomito,
vertigini, instabilità e mal di testa, e la diagnosi clinica si basa su esame
neurologico concentrati e un indice ragionevole di sospetto. Risultati
dell'esame sono incoordinazione, atassia e nistagmo orizzontale. I
pazienti possono anche presentare con stato di coscienza alterato o coma.
Caratteristiche
radiografiche
CT
Caratteristiche
tipiche di infarto, come la perdita precoce di grigio-bianco differenziazione,
hypoattenuation ed edema, progressione di encefalomalacia cronica possono essere
identificati nei rispettivi territori vascolari.
Trattamento
e la prognosi
La
mortalità correlata ad infarti cerebellari è superiore a quella di altri
territori vascolari.Questo è generalmente dovuto alla concomitante infarto del
tronco encefalico, o idrocefalo compressione, piuttosto che infarto cerebellare
in sé. Complicanze insoliti includono:
Cause di ictus del tronco encefalico e
cervelletto - Nozioni di base
Gli
Strokes possono essere ischemici (mancanza di flusso di sangue), o emorragici
(fuoriuscita di sangue nel cervello). I fattori di rischio per l'ictus sono
considerati qui.
Ictus
ischemici sono causati da ostruzione dei vasi sanguigni. I blocchi possono
provenire da una sorgente distante - come un coagulo dal cuore , che sono
chiamati "emboli". I blocchi possono derivare da coagulazione
dall'interno dei vasi. Allora sono chiamati "trombi". Gli Ictus
trombotici sono più spesso attribuite ad accumulo di colesterolo all'interno
delle pareti dei vasi sanguigni, producendo una turbolenza e irruvidimento
della parete, su cui si forma un coagulo. Un'altra causa comune di ictus
trombotico è un calo sostenuto della pressione sanguigna - come potrebbe essere
causa di un arresto cardiaco per alcuni minuti. Ci sono fonti solo occasionali
di ictus ischemico di fuori di questi generali due categorie (emboli e trombi).
Ad esempio, possono essere causati da ostruzione meccanica dei vasi sanguigni,
come potrebbe accadere durante una manipolazione chiropratica del collo ad
alta velocità , o qualche altro evento che provoca un movimento del collo molto
energica. Abbiamo incontrato i pazienti che hanno subito ictus (ad esempio),
dopo montagne russe.
Ictus
emorragico - perdita di sangue nel cervello - -Sono più comunemente causata da
pressione sanguigna troppo alta. Altre possibili cause sono irregolarità nella
coagulazione del sangue, danni alle pareti dei vasi sanguigni di diversi
processi (tra cui ictus ischemico). Nel tronco encefalico, il circuito è
stretto e gli ictus emorragici sono spesso devastanti.
MALATTIA
DEI PICCOLI VASI: PICA AICA SINDROME DELL'ARTERIA LABIRINTICA SCA
Cause di ictus del tronco encefalico - Le basi
l’ictus (Stroke) può essere ischemico (mancanza
di flusso sanguigno) o emorragico (fuoriuscita di sangue nel
cervello).
FATTORI
DI RISCHIO PER TIA E L’ICTUS
To some extent, one can predict risk of stroke. In una certa misura, si può predire
il rischio di ictus. Risk factors that are well
known include: I fattori di rischio che sono ben noti includono:
High Blood Pressure (2)
Alta pressione sanguigna (2)
Collagen Vascular disease Malattie vascolari del collagene
Heart problem such as atrial fibrillation (1.5) or old
infarction (2.7)
Problema di cuore come la fibrillazione atriale (1.5) o vecchio infarto
(2,7)
**Numbers in () are derived from Whisnant et al, 1996 ** I numeri in () sono derivati
da Whisnant et al, 1996 Rischio di pressione sanguigna elevata è
ripida e chiaro.
Rischio dovuto alla pressione
sanguigna. Risk from elevated blood pressure is
steep and clear.Ad esempio, nello studio UK TIA, il rischio di recidiva
di ictus è aumentato del 28% per ogni aumento di 10 mm Hg della pressione
sistolica tra 130 e 160 (Farrell et al, 1991).
Although LDL-cholesteral,
HDL cholesteral seems to reduce risk of stroke (Sacco et al, 2001). Anche se LDL-colesterolo, HDL
colesterolo sembra ridurre il rischio di ictus (Sacco et al, 2001). If your HDL cholesterol is > 35, then subtract one risk
factor (a negative risk factor). Se il colesterolo HDL è> 35, bisogna
sottrarre un fattore di rischio (un fattore di rischio negativo). Mitral valve prolapse is not a significant risk factor,
overall (0.8 risk). Il prolasso della valvola mitrale non è un fattore
di rischio significativo, nel complesso (rischio x 0,8). TIA is a very strong risk factor for stroke (5.6 x risk).
TIA è un fattore di rischio molto forte per l'ictus (rischio x 5.6). In general, relative risk for most of the above factors
decreases with age (Whisnant et al, 1996), lending support for a unaggressive
approach to risk factors in individuals of advanced age. In generale, il
rischio relativo per la maggior parte dei fattori di cui sopra diminuisce con
l'età (Whisnant et al, 1996), sostenendo un approccio non aggressivo per i
fattori di rischio in individui di età avanzata.
Risk from cholesterol can
be further stratified into three groups, based on LDL (total cholesterol -
HDL)-(triglycerides).
Rischio da colesterolo può essere ulteriormente stratificata in tre gruppi,
sulla base LDL (colesterolo totale - HDL) - (trigliceridi).
LDLLDL
Risk FactorsFattori di rischio
Risk Level for vascular diseaseLivello
di rischio per malattia vascolare
< 130 <130
None Nessuno
Low Basso
130-159 130-159
Less than 2 Meno di 2
Moderate Moderato
>130 > 130
more than 2 più di 2
High Alto
Controllable risk factors
include being overweight, having high (> 140/90) or low blood pressure,
heart disease, diabetes and smoking.
Fattori di rischio controllabili comprendono il sovrappeso, con elevata (>
140/90) o bassa pressione sanguigna, malattie cardiache, diabete e fumo. Atrial fibrillation is a particularly important risk
factor -- stroke occurs in 4.5% of untreated patients with atrial fibrillation
per year. La fibrillazione atriale è un fattore di rischio
particolarmente importante – l’ictus si verifica nel 4,5% dei pazienti trattati
con fibrillazione atriale all'anno. While uncommon,
chiropractic neck manipulations can cause compression or tears of the vertebral
arteries (Vibert et al, 1993; Smith et al, 2003), and for this reason,
maneuvers involving neck "cracking" should be specifically avoided in
individuals with vertigo. Mentre , le manipolazioni chiropratiche del
collo possono causare compressione o rotture delle arterie vertebrali (Vibert
et al, 1993; Smith et al, 2003) e per questo motivo, le manovre
"cracking" che coinvolgono il collo devono essere specificamente
evitati nei soggetti con vertigine. Whiplash
injuries can also damage the vertebral arteries as the arteries traverse the
vertebrae of the neck. Il colpo di frusta può anche danneggiare le
arterie vertebrali che passano attraversano le vertebre del collo.
L’ictus ischemico è causato da
ostruzione dei vasi sanguigni. L’ostruzione può provenire da una sorgente
distante - come ad esempio un coagulo dal cuore – che poi viene chiamato
"embolo". I blocchi possono derivare da coagulazione
dall'interno dei vasi - che poi sono chiamati "trombi". Gli ictus
trombotici sono più spesso attribuiti a accumulo di colesterolo all'interno
delle pareti del vaso sanguigno, che producono turbolenza e ispessimento
irregolare della parete, con successiva e conseguente formazione di coaguli.
Un'altra causa comune di ictus trombotico è una caduta di pressione sanguigna
- come potrebbe essere ad esempio causata da un arresto cardiaco di alcuni
minuti. Al di fuori di queste due categorie generali (emboli e trombi)ci
possono essere solo fonti occasionali di ictus ischemico Ad esempio, causati da
ostruzione meccanica dei vasi sanguigni – come potrebbe accadere durante una manipolazione chiropratica del collo ad alta
velocità o qualche altro evento che causa un movimento collo molto
forte. Si sono riscontrati (ad esempio), ictus in pazienti subito dopo
che gli stessi erano stati sulle montagne russe.
Ictus emorragici - fuoriuscita di
sangue all'interno del cervello - sono più comunemente causate da pressione
sanguigna troppo alta. Altre possibili cause comprendono irregolarità
nella coagulazione del sangue, danni alle pareti dei vasi sanguigni da vari
processi (compreso l'ictus ischemico). Nel tronco encefalico, il circuito
è stretto e gli ictus emorragici sono spesso devastanti.
La sindrome PICA è
conosciuto anche come "sindrome laterale midollare", o "sindrome
di Wallenberg", dopo la descrizione di Wallenberg nel 1895. Questo è il
colpo del tronco cerebrale più comune. Vedere questo link per
maggiori dettagli.
AICA (sindrome dell’arteria cerebellare antero inferiore).
La sindrome AICA è di
solito accompagnata da vertigini e sordità omolaterale unilaterale dalla
labirintica ischemia dell'arteria. Si tratta di un ictus del tronco
cerebrale comune. Vedere questo link per
maggiori dettagli.
Sindrome dell'arteria labirintica
L'arteria uditiva
labirintico o interno di solito prende la sua origine da AICA, ma può anche
prendere origine dal PICA o l'arteria basilare. Fornisce l'orecchio
interno. Nel canale uditivo interno o IAC fornisce ganglio di
Scarpa. Dopo l'uscita si divide in arteria cocleare comune e anteriore
arteria vestibolare. L'arteria cocleare comune ulteriore divide in arteria
cocleare e l'arteria vestibolococleare, quest'ultima formando posteriore
arteria vestibolare e il ramo vestibolare. La principale arteria cocleare
fornisce il apicale 3/4 della coclea, e il ramo cocleare, le basali 1/4 (alte
frequenze). L'arteria vestibolare posteriore fornisce il sacculo inferiore
e l'ampolla del SCC posteriori. L'arteria vestibolare anteriore è una arteria
minore che fornisce utricolo, sacculo superiore, ed ampolla anteriore e nei
canali semicircolari laterali (Kim et al, 1999). L'arteria labirintica è
un arteria sottile terminale e come tale può essere relativamente più
vulnerabile di altre circolazioni.
La diagnosi può essere
difficile perché il cervello può mostrare nessuna lesione. Questo è un
piccolo vaso sanguigno e studi di imaging come TAC-angiografica o RMA
perdere. Il contrasto è assolutamente necessario (durante MRA).
I sintomi principali sono
atassie cerebellari omolaterali (peduncoli cerebellari medi e / o superiori), nausea
e vomito, impastata (pseudobulbare) discorso, la perdita del dolore e della
temperatura sul lato opposto del corpo. Sordità parziale, tremore degli
arti superiori, una sindrome di Horner omolaterale e mioclono palatale sono stati
segnalati. Clinicamente, questo colpo può essere impossibile da
distinguere da una AICA parziale o ictus territorio PICA. E 'molto più
raro di uno dei due. Pulsione oculare lontano dal lato della lesione è
stata riportata nella sindrome SCA. Diagnosi della corsa avviene tramite
risonanza magnetica.
Nella sindrome di Weber,
vi è un danno per il mesencefalo, compresi i fascicoli del III° nervo
all'interno del mesencefalo e anche lì vi è un danno al fascio piramidale prima
della decussatione (area del peduncolo cerebrale). Ciò si traduce in una
paralisi del III° nervo omolaterale combinata con una emiplegia
controlaterale. Questa sindrome può derivare da danni alla SCA.
Sindrome
di Weber
Infarto del mesencefalo
laterale risultante da un aneurisma dell'arteria cerebellare
superiore. La lesione è qui solo posteriormente al peduncolo cerebrale.
Infarto Mediale Midollare (sindrome di Dejerine).
0,5% di tutte infarti cerebrali. Emiparesi controlaterale
risparmiando il volto, perdita hemisensory del tipo colonna posteriore
(controlaterale). Debolezza della lingua è omolaterale al
infarto. Patologia può essere in arteria vertebrale o di un arto mesiale
dell'arteria vertebrale dopo PICA, può verificarsinistagmo Upbeat . Malattia dei
piccoli vasi (diabete, ipertensione, ipercolesterolemia) è la causa usuale.
Caplan LR. Vertebrobasilar
disease. In Barnet HJM (and others, Eds), Stroke: Pathophysiology,
Diagnosis and Management. New
York: Chrchill-Livingstone, pp 549-619, 1986
Faught E, Oh SJ. Brainstem
auditory responses in brainstem infarction. Stroke 1985. 16:701-705
Fisher CM. Vertigo in
cerebrovascular disease. Arch Otolaryngol 1967;85:529-534
Grad A, Baloh RW. Vertigo of
vascular origin: clinical and electronystagmographic features in 84 cases.
Arch Neurol
1989;46;281-284
·Kerber
KA, Brown DL, Lisabeth LD, et al. Stroke among patients with dizziness,
vertigo, and imbalance in the emergency department: a population-based
study. Stroke. 2006; 37 :2484–7. [ PMC free article ][ PubMed ]
Kim JS and others. Internal
auditory artery infarction. Neurology
1999:42:40-44
Kubik CS, Adams RD. Occlusion of
the basilar artery: a clinical and pathological study. Brain 1946: 69:73-121
Kwa VIH, Zaal LH,
Verbeeten B, Stam J. Disequilibrium in patients with atherosclerosis. Relevance of pontine
ischemic infarction. Neurology 1998;51:570-573.
Savitz SI, Caplan LR, Edlow
JA. Pitfalls in the diagnosis of cerebellar infarction. Acad
Emerg Med. 2007;14 :63–8. [ PubMed ]
La sindrome PICA è
conosciuto anche come "sindrome laterale midollare", o
"sindrome di Wallenberg"o “sindrome
dell’arteria cerebellare postero-inferiore”, dopo la descrizione di
Wallenberg nel 1895.Dipende da un’occlusione o dell’arteria cerebellare
posteroinferiore (PICA) o del tratto terminale di un’arteria vertebrale da
dove questo ramo origina. La lesione ischemica interessa il segmento
dorso-laterale del bulbo (Fig. 4A-B). Questo è l’ictus più comune del
tronco cerebrale.
Fig. 4- A) sede della lesione
infartuale defla 5. ischemica lat. del bulbo (s. di Wallemberg) (da Baloh R.W.,
1984); B) sez. trasversa del bulbo che illustra la distribuzione dell’a.
cerebellare post. inf. (da Silver F.L. in Sharpe J. e Barber H. Ed., 1993);C)
sez. trasversa della zona dorso laterale bulbo-pontina irrorata dall’a.
cerebellare ant.-inf. (da Silver F. L. in Sharpe J. e Barber H. Ed., 1993). da Megighian
Otoneurologia Piccin2008
SINTOMATOLOGIA
La sintomatologia compare all’improvviso ed è caratterizzata da
vertigine assai intensa, da perdita di equilibrio con latero-pulsione sul lato
della lesione (emiatassia omolaterale), da disartria, ptosi e miosi, da cefalea
occipitale e da sintomi neurovegetativi ipervagotonici..All’inizio, l’episodio vertiginoso può venir confuso con quello di
una lesione acuta del labirinto o delle strutture nervose che appartengono
all’arco primario vestibolare. Il nistagmo può essere unico, di tipo
orizzontale, in genere diretto verso il lato opposto alla sede della lesione
oppure multiplo. In tal caso, al nistagmo “paralitico” si associa un nistagmo
da “direzione dello sguardo”, prevalentemente orizzontale- rotatorio o
rotatorio, diretto verso il lato del focolaio ischemico. Compaiono ampi
movimenti sacca- dici (ipermetrici), sia volontari che involontari, diretti
verso il lato della lesione mentre quelli diretti verso il lato opposto sono
molto piccoli. Anche i movimenti oculari lenti di conduzione (pursuit) appaiono
asimmetrici. Oltre ai segni di sofferenza cerebellare: asinergia, atassia,
adiadococinesi (per l’interessamento del distretto postero-inferiore
dell’emisfero cerebellare e del verme), si osservano disfonia e disfagia per
paralisi omolaterale del X e del 1X, ipoestesia dell’emifaccia con riduzione o
scomparsa del riflesso corneale per interessamento del nucleo e della radice
discendente del V, paralisi omolaterale del VII. Frequente è la comparsa della
sindrome di Claude Bernard-Horner (miosi, enoftalmo, ptosi palpebrale)
omolaterale alla sede della lesione per interessamento delle vie simpatiche
oculomotorie che corrono in questo distretto bulbare. Nel lato opposto alla
lesione, si osserva una emianestesia dissociata termo-dolorifica.
Il nistagmo provocato da stimolo calorico presenta, oltre alle
modificazioni qualitative delle scosse (prevalenza della fase lenta, aritmia, variazioni
della forma), un’alterazione dei valori dei parametri quantitativi che varia in
rapporto all’estensione della lesione vascolare e, soprattutto, al grado di
interessamento dell’area nucleare vestibolare o dei distretti nervosi
iuxtanucleari. Nella sindrome di Wallemberg, come in altre lesioni che
interessano il segmento del bulbo posteriore all’oliva, non è possibile mettere
in evidenza, nel corso della reazione vestibolare, la deviazione tonica degli
arti superiori (prova di Bàràny) per l’interessamento delle vie di connessione
vestibolo-spinali. Nel corso della reazione vestibolo-oculomotoria provocata
dallo stimolo calorico, non si osserva la comparsa del “perverted nystagmus”.
Le scosse del NOC presentano alcune alterazioni morfologiche
legate all’ interferenza del nistagmo spontaneo o del nistagmo da “direzione
dello sguardo” e alla presenza di anomalie dei saccadici soprattutto di quelli
diretti verso il lato della lesione.
La maggior parte dei pazienti dopo questo ictus recupera
molto bene e spesso riprendono le loro precedenti attività (Nelles et al,
1998). I pazienti spesso hanno la sindrome di Horner (ptosi monolaterale,
miosi e anidrosi facciale). Ci possono essere anche saccadici dismetrici
(overshoot), pulsione saccadici (tirando dell'occhio durante le saccadi
verticali verso il lato di lesione).La prognosi è generalmente molto buono con
recupero pieno o quasi completo atteso entro i 6 mesi. La diagnosi si fa
generalmente con la risonanza magnetica. CT-angiografia con ricostruzione
in 3D ha ottenuto abbastanza buono in questi ultimi anni per essere utile
troppo.
L’esame ABR è spesso
anormale nelle persone con una sindrome di Horner centrale (Faught e Oh, 1985),
ma siccome la lesione nella sindrome di Wallenberg è abitualmente inferiore
alle vie uditive, la sindrome di Horner prodotta dalla Wallenberg generalmente
non è associata ad ABR anormali.
La PICA deriva
normalmente dall'arteria vertebrale , oppure da un ramo distinto dell'arteria
basilare. A causa dell'origine molto più comune dalla arteria vertebrale,
la maggior parte delle sindrome (ictus) "PICA" in realtà sono dovuti
ad occlusione dell'arteria vertebrale (Kim 2003) l’embolia cardiaca provoca
solo il 5% di questi ictus , mentre la dissezione provoca il 15% (Kim, 2003).
PICA è il sito più comune
di occlusione dalla moltiplicazione dei trombi o embolie causata da lesioni
alla terza sezione dell'arteria vertebrale, e la sindrome di Wallenberg è la
patologia più comune causata da manipolazione chiropratica (Caplan, 1986).
References
·Caplan LR. Vertebrobasilar disease. In Barnet HJM (and others,
Eds), Stroke: Pathophysiology, Diagnosis and Management. New York: Chrchill-Livingstone,
pp 549-619, 1986
·Faught E, Oh SJ. Brainstem auditory responses in brainstem
infarction. Stroke
1985. 16:701-705
L'arteria cerebellare antero inferiore(AICA) è
un'arteria nel cervello che fornisce parte del cervelletto .
Esso deriva dalla arteria basilare a livello della giunzione tra
il midollo allungato ed i ponte nel tronco cerebrale . Si passa all'indietro da
distribuire alla parte anteriore della superficie inferiore del cervelletto,
anastomosi con il posteriore inferiore cerebellare ramo dell'arteria vertebrale
. Fornisce il quartiere inferiore anteriore del cervelletto.
Dà anche origine, nella maggior parte dei casi dell'arteria
labirintica; Tuttavia, in altri casi l'arteria labirintica può emergere come un
ramo della basilare. La sindrome AICA solito si presenta con vertigini e
sordità omolaterale unilaterale dalla labirintica ischemia dell'arteria. I
grandi ictus sono accompagnati da paralisi facciale omolaterale e atassia. E '
per frequenza il secondo più comune ictus del tronco cerebrale, circa il 10%
degli ictus colpiscono la PICA.
L'entità di questo tratto
è estremamente variabile. Sintomi simili a quelli della malattia di Meniere (udito fluttuante,
acufeni, vertigini) possono essere causati anche da TIA in tale distribuzione
(Lee e Cho, 2003). La bilateralità delle fluttuazione uditive suggerisce
una causa vascolare, ma la maggior parte degli ictus da AICA si presentono con
sintomi uditivi unilaterali. L'AICA ha anche una origine molto variabile e
può prendere origine dalla porzione caudale mediana del ponte. Gli ictus
del territorio dell’AICA possono presentare anche solo vertigini . La
diagnosi si ottiene generalmente tramite risonanza magnetica.
References
·Lee H, Cho YW. Auditory disturbance as a prodrome of anterior
inferior cerebellar artery infarction. J. Neurol Neurosurg Psych 2003:74:1644-1648
Tab. I - Sintomi e strutture nervose
interessate nelle lesioni infartuali nel territorio irrorato dalla P1CA e da AICA
Sintomi
Infarto nel
territorio
della PICA
Infarto nel
territorio
della AICA
Vertigine
Nn. vest., distretto
cereb. post-inf.
Distretto dorsolat.
b-pontino, flocculo, labirinto
Ipoacusia, acuf.,
vertigi- ne
Assenti
Ipoacusia, acuf.,
vertigine
Segni di atas sia -
Presenti (tratto cereb.
ventrale)
Presenti (ped cereb.
medio).
Disfagia
I X
Assente
Disfonia
X
Emianest. fac- ciale
omolat
V
V
Paralisi del
Presente
Presente
Emi-ipoeste- sia
controlat
Presente (tratto
spino-tal.)
Presente (tratto
spino-tal.)
S di Bernard-Horner
Fibre simp. disendent.
Fibre simp. disendent.
APPROFONDIMENTO
La differenziazione
clinica di infarto cerebellare da comuni Vertigo Sindromi
Questo articolo riassume
l'approccio pronto soccorso a diagnosticare infarto cerebellare nel paziente
con vertigini. Vertigo è definito e l'identificazione di una sindrome
vertigini è discusso. La differenziazione delle sindromi vertigine comuni
come benigna vertigine parossistica posizionale, la malattia di Meniere,
vertigini emicranica e neurite vestibolare è riassunta. Conferma di una
sindrome vertigini periferica riduce sostanzialmente il rischio di infarto
cerebellare, così come indicatori di un disturbo periferica quale un test
impulso testa anormale. Circa il 10% dei pazienti con infarto cerebellare
presente con vertigini e senza localizzative deficit neurologici. La
maggior parte di questi può avere altri segni di vertigini centrale, in
particolare in direzione cambia nistagmo e grave atassia.
INTRODUZIONE
Mentre la maggior parte
dei pazienti che si presentano per i servizi di emergenza (ED) con isolate
vertigini hanno disturbi benigni, circa il 0,7-3% avere infarto
cerebellare. 1,2 perché i sintomi di
infarto cerebellare sovrapposizione sostanziale con le condizioni benigne è
comunemente trascurato, con un tasso di diagnosi errate stimata al 35% 2. I pazienti con
infarto cerebellare perso in generale sono a più alto rischio di complicanze,
con un tasso di mortalità forse pari al 40%. 3
Diagnosi fisica è la
modalità diagnostica più importante per infarto cerebellare. Il ricorso a
tomografia computerizzata (CT) non è sufficiente, perché è solo il 26%
sensibile per ictus acuto. 4, invece, importanti
segni fisici sono presenti nella maggior parte dei pazienti con infarto
cerebellare.
Questa revisione prima
affronta la differenziazione di infarto cerebellare dai quattro sindromi
vertigini più comuni:. Vertigine parossistica posizionale benigna (VPPB), la
malattia di Meniere, vertigini emicranica e neurite vestibolare 5 Poi ci rivedrà la
diagnosi di infarto cerebellare fisico. Infine, vi proporremo le
indicazioni per neuroimaging.
Definizione di Vertigo
La Vertigine è definita
come una illusione patologica di movimento. 6 Più comunemente
sperimentato come sensazione di rotazione, esso deriva da uno squilibrio
patologico nel sistema vestibolare periferico o centrale. I pazienti
spesso semplicemente relazionano sensazione di vertigini, e un ulteriore
interrogatorio è necessario per identificare le vertigini. 7 Si raccomanda cautela
nella classificazione del paziente vertigini. Mentre si differenziano le vertigini
da uno disequilibrio, presincope e stordimento tradizionale , gli studi
dimostrano che i pazienti utilizzano termini sovrapposti per descrivere la loro
esperienza e possono anche cambiare idea durante un singolo incontro
clinico. 8 Il modo migliore per
un medico per identificare una sindrome vertigine è quello di realizzare che
mentre la maggior parte dei pazienti riferisce le classiche vertigini rotazionali,
circa il 17% non lo faranno. 9 Questi pazienti
possono segnalare episodica disturbi dell’equilibrio o vertigini che peggiorano
con il movimento della testa.
Vertigine parossistica
posizionale benigna
La Vertigine Parossistica
Posizionale Benigna è una condizione distinta e che in genere non si confondere
con l’infarto cerebellare. Sono stati descritti disturbi centrali della
VPPB, ma tendono ad essere causati da tumori, piuttosto che d ictus e sono
riconosciuti dall'associazione con altre anomalie. 10 I pazienti
presentano brevi episodi di vertigini intense, che peggiorano con i cambiamenti
di posizione. I parossismi con sintomi intensi che durano meno di un
minuto vengono definiti come una provocazione posizionale. I pazienti stanno
bene, fino a quando un movimento della testa, generalmente verticale, precipita
il parossismo dei sintomi. Nella maggior parte dei casi la VPPB è
idiopatica, anche se il 10% segue un attacco di neurite vestibolare e il 20% a
seguito di un episodio di trauma cranico. 11
La forma più comune di
VPPB, è causata dagli otoliti nel canale semicircolare posteriore, viene
diagnosticata trovando nistagmo torsionale nel test di Dix-Hallpike. 12 L'esaminatore tiene
la testa del paziente seduto a 45 ° a sinistra o a destra. Ciò allinea il
canale semicircolare posteriore sul piano verticale. Il paziente deve
essere invitato a tenere il braccio sull'esaminatore per la
stabilità. L'esaminatore poi porta il paziente nella posizione supina, con
la testa penzoloni dalla barella di 10 ° -30 °. Ciò causa una grande
rotazione del canale semicircolare posteriore nel proprio piano, spostando il
otoliti liberi che riproducono i sintomi. Il test è considerato positivo
se si provoca la torsione nistagmica caratteristica ed un nistagmo
verticale. 13
La sensibilità della
manovra Dix-Hallpike per VPPB non è stata ben descritta nell'impostazione
. Anche senza un test Dix-Hallpike positivo, i pazienti con BPPV possono
essere differenziati da quelli con infarto cerebellare dai loro sintomi
episodici e dalla posizione. Coloro che non si adattano a questa
descrizione richiedono considerazione diagnostiche alternative.
Malattia di Meniere
La Malattia di Meniere è
sospettata nel paziente che si presenta con simultanee vertigini e disturbi cocleari.
I disturbi cocleari possono essere perdita uditiva, acufeni o senso di
orecchio pieno. Chiamata anche idrope endolinfatica, si pensa che la malattia
di Meniere è causata da un accumulo di liquido endolinfatico nell'orecchio
interno. Gli episodi comunemente durano poche ore, anche se possono
variare da 20 minuti ad un paio di giorni. La diagnosi formale richiede una
perdita dell'udito documentata all'esame audiologico in almeno una
occasione. 14 I pazienti possono
avere esame audiologico normale tra un episodio e l’altro.
Non è comune la comparsa
di vertigini isolate e la perdita di udito, anche se esistono casi
clinici. 15 Questa si verifica
solo nel 0,3% di tutti gli infarti del tronco encefalico e tende a presentarsi
con sordità completa omolaterale. 16 In generale, le vertigini
con perdita di udito , a meno che non ci sia sordità totale omolaterale,
indicano un disturbo periferico. Il medico deve eseguire un esame
neurologico completo e neuro-otologica in questi pazienti, come discusso di
seguito. I pazienti con un negativo work-up per l'ictus dovrebbero essere
riferiti ad un otorinolaringoiatra per ulteriori test e prendere in considerazione
altre diagnosi, come la labirintite e lo schwannoma. 6,17 I pazienti con
malattia di Meniere tendono ad avere episodi ricorrenti, per cui se il paziente
non ha avuto precedenti episodi, deve essere sospettata invece una labirintite. Si
pensa che la Labirintite abbia lo stesso processo eztiologico della neurite
vestibolare, con l’ulteriore coinvolgimento del sistema uditivo.
Vertigine Emicranica
Nonostante sia la seconda
causa più comune di vertigine osservata nella pratica clinica, la vertigine
emicranica rimane sotto-riconosciuta. 5 Il suo spettro clinico
può essere elusiva. La metà dei pazienti si presenta senza cefalea. 18 Le sue
caratteristiche di presentazione possono variare, non solo tra i differenti
pazienti, ma nello stesso paziente nel tempo. Alcuni hanno aura, mentre
altri non la presentano . Alcuni hanno fotofobia durante gli attacchi,
mentre altri non la presentano. Per alcuni, la vertigine emicranica
sembra essere come un'aura che dura pochi minuti (18%), ma per gli altri la
vertigine dura per più di 24 ore (27%). 19 L'esame fisico
dovrebbe rivelare un normale esame neurologico, tra cui il coordinamento e
l'andatura. La Video oculografia ha dimostrato risultati di nistagmo
centrale,. 20
Sono stati proposti
criteri diagnostici formali per vertigine emicranica. 21 criteri rigorosi
richiedono: 1) episodi ricorrenti di vertigine; 2) una diagnosi di
emicrania formale da International Headache Society (IHS) criteri;3) un sintomo
dell'emicrania durante l'attacco (ad esempio l'emicrania, con fotofobia, o
aura); e 4) l'esclusione di altre cause. La categoria di " probabile
vertigine emicranica " viene usato per i pazienti con alcuni elementi
nella presentazione di suggerire l'emicrania, ma nessuna altra causa
identificabile.
L’Infarto cerebellare non SI
dovrebbe presentare con I sintomi associatI di emicrania, quindi la maggior
parte dei pazienti con criteri di vertigine emicranica e CON un normale esame
neurologico possono essere trattati per il loro processo di emicrania senza
ulteriore work-up.
Neurite Vestibolare
La Neurite vestibolare è
caratterizzata dalla sindrome vestibolare acuta, causata da tono vestibolare
diminuita su di un lato. Attualmente si pensa che l'eziologia sia virale. 22 La neurite vestibolare
si riconosce dai reperti caratteristici sull’anamnesi e con l'esame vestibolare. La
storia rivela in genere un inizio graduale, a differenza dell’ ictus in cui i
sintomi raggiungono la massima intensità all’ esordio. Nella neurite
vestibolare, i sintomi di picco si hanno durante il primo giorno e cominciano a
migliorare nel giro di pochi giorni, anche se il pieno recupero richiede
settimane o mesi. 6 La vertigine
associata è continua e costante, anche se come tutte le forme di vertigine, è
caratteristica l’esacerbazione posizionale, in quanto ogni movimento della
testa amplifica la differenza del tono vestibolare bilaterale. I sintomi neurovegetativi
associati di nausea e vomito sono prominenti.
L’Esame generale
neurologico è normale, tra cui i riflessi motori, sensoriali, i nervi cranici e
lo stato mentale. Ancora più importante, il coordinamento degli arti è
conservato nella prova dito-naso, e le prove di movimento a rapida alternanza tallone-piede
. Anche se è prevista qualche lieve incoordinazione, il paziente deve
mantenere la capacità di deambulare. Il loro sistema nervoso centrale
continua a integrare la sue informazioni somatosensoriali, visive e
propriocettive, coordinando la deambulazione.
Il paziente vestibolopatico
ha il tono vestibolare diminuito sul lato interessato. Questo squilibrio del
tono normalmente si verifica quando il paziente ruota la testa da quel lato,
in modo che il paziente vestibolopatico sperimenta come se il mondo continua a
girare quando ruota la testa sul lato colpito. Il riflesso
vestibolo-oculare provoca una lenta deriva di compensazione degli occhi verso
l'orecchio ipoattivo, che è la fase lenta del nistagmo. La fase veloce del
nistagmo è il battito correttivo opposta all'orecchio ipoattivo. 1 La sua direzione è
orizzontale e torsionale, ed è considerato unidirezionale, cioè indipendentemente
da dove il paziente guarda, la direzione del nistagmo non cambierà. 22 è accentuato quando
guarda lontano dall'orecchio ipoattivo (Legge di Alexander). Il paziente
sente questo e spesso chiude gli occhi quando distogliere lo sguardo dal lato
ipoattivo. Va ricordato che il nistagmo unidirezionale può verificarsi
anche nel 46% dei pazienti con infarto cerebellare quindi non può essere usato
per confermare un disturbo periferico. 23 Tuttavia, vi è una
constatazione fisica in particolare, che può fornire un aiuto.
Il riflesso vestibolo-oculare
(VOR) è una cerniera tra il movimento degli occhi per il movimento della
testa. Quando una persona normale guarda lateralmente, il labirinto di
quel lato segnala la rotazione e gli occhi si muovono automaticamente in
direzione opposta per mantenere lo sguardo fisso. Ad esempio, il lettore
può ruotare la testa alternativamente per ogni lato e continuare ancora a
leggere, a causa del VOR. Per il paziente vestibolopatico, quando la testa
viene rapidamente spostata verso il lato patologico, gli occhi si muovono con
la testa e la fissazione visiva viene interotta. Un saccade di rifissazione
("catch-up") si vede quando il paziente guarda indietro l'oggetto
originale. Questo è testato dal test, chiamata anche la prova di impuls
test. 24 La testa del
paziente viene posto sulla linea mediana, e il paziente viene istruito a
mantenere la fissazione visiva sul naso dell'esaminatore. L'esaminatore
spinge la testa del paziente rapidamente da un lato, e se si vede un saccade di
rifissazione indietro sul naso , è presente una paresi del canale sul lato
verso cui la testa viene girata (Figura
1). La
prova deve essere eseguita su entrambi i lati. Un video del test in corso
di esecuzione è disponibile online. 25,26 In altre
impostazioni che è stato descritto come sensibile solo nel 34-39% di
ipofunzione vestibolare. 27,28 Quando studiato in
popolazioni al pronto soccorso più altamente selezionati un test positivo di impuls
test è stata % sensibile nel 100 per un disturbo periferico. 26 Un test impulso test
negativo è stato trovato dal 91 al 96% dei pazienti con infarto
cerebellare. 23,26 Ulteriori studi
devono affrontare il ruolo del test impulsi testa in pazienti con sindromi ED
indifferenziate vertigini. Fino a quel momento si può solo dire che un
impuls test positivo è più coerente con una vestibulopatia periferica acuta,
come la neurite vestibolare, e un impuls test negativo fa pensare ad un
infarto cerebellare.
Impulse
test A: L'orecchio destro ha intatta la funzione vestibolare
periferica. Quando la testa è girata a destra, il riflesso
vestibolo-oculare sposta gli occhi per mantenere fissazione visiva. B:
L'orecchio destro ora è compromessa funzione vestibolare. Quando la...
Infarto cerebellare
L’Infarto cerebellare
rappresenta circa il 2,3% degli infarti acuti generale. 29, che possono derivare
da un'occlusione dell'arteria cerebellare superiore (SCA), dell’arteria
cerebellare antero inferiore (AICA), o dell’arteria cerebellare posteriore
inferiore (PICA). Gli infarti cerebellari più grandi producono sintomi e
segni che localizzano al tronco encefalico, come diplopia, disartria, arti
atassia, disfagia, e ipotonia o parestesia. Circa il 10% dei pazienti con
infarto cerebellare si può presentare con vertigini isolate, cioè, vertigini senza
altri sintomi come interessamenti motori, sensoriali, dei riflessi, nervi
cranici, o negli esami di coordinazione degli arti. La maggior parte di
questi sono infarti del ramo mediale del PICA (96%). 23
I pazienti con vertigini isolate
a causa di un infarto cerebellare rappresentano una sfida diagnostica
significativo al medico del pronto soccorso emergenza (EP). E’ noto per sia
spesso mal diagnosticato, con conseguente successiva disabilità.3 Mentre la sua rarità
osta buoni studi DE sui suoi segni che presentano, la letteratura è sufficiente
per offrire alcuni indizi storici e fisici che possono allertare il clinico sulla
possibilità di un infarto cerebellare.
Primo, l’ictus in generale
tende a presentare un'insorgenza improvvisa e con sintomi immediati , che di
solito raggiungono la massima intensità immediatamente. In secondo luogo, i
fattori di rischio vascolare aumentare la probabilità a priori di malattia. L’ ipertensione
e le malattie cardioaortiche si trovano nella maggior parte dei pazienti con
infarto cerebellare, e una fonte embolica si è trovato nel 24-40%. 23,29, infine, due segni
fisici facilmente trascurati si è dimostrato che indicano l’infarto
cerebellare.
Il primo è l'atassia
grave, che è stato classicamente considerato un segno di vertigini
centrale. 6 Il settanta per
cento dei pazienti con infarto cerebellare e vertigini isolate si presenterà
con incapacità di camminare senza sostegno. 23 Graded da due
osservatori indipendenti, questo sembra essere un riscontro oggettivo e
riproducibile, anche se gli osservatori non sono stati accecati per la
diagnosi. L'altro 29% ha uno squilibrio da lieve a moderato con presenza di
deambulazione, che di per sé non sarebbe capace di consentire la
differenziazione dalla neurite vestibolare.
Il secondo importante
segno fisico di infarto cerebellare è il nistagmo con direzione mutevole,
chiamato anche nistagmo multidirezionale, o nistagmo evocato dallo sguardo. Questi
pazienti hanno nistagmo che cambia direzione secondo lo sguardo del
paziente. Ad esempio, se il paziente guarda verso destra batte verso
destra, e quando l'aspetto paziente lasciata batte verso sinistra. Questo
è risultato essere el 56% sensibile per l’ infarto cerebellare, anche se i
medici non sono stati accecati alla diagnosi. 23 Il medico dovrebbe
evitare l'errore di cercare il nistagmo con lo sguardo portato in posizione estrema
laterale, in quanto si produce un nistagmo end-point, che è normale e riflette
solo fatica muscolare .30 Alcuni farmaci, in
particolare gli anti-epilettici e l'alcool, possono pure causare nistagmo.
L'incapacità di camminare
senza sostegno e il nistagmo che cambia direzione sono due segni importanti
perché sono comunemente presenti anche quando non sono presenti altri risultati
del tronco encefalico ischemia. Almeno uno di questi due segni è stato visto nell
84% (21 su 25) dei pazienti con infarto cerebellare e vertigini isolate. 23
Indicazioni per
Neuroimaging
Non sono state stabilite
raccomandazioni basate sull'evidenza per neuroimaging nel paziente vertiginoso.
Sulla base dei dati attualmente disponibili, chiare indicazioni per
neuroimaging includono alcun deficit neurologici focale, l'incapacità di
camminare senza sostegno e la direzione mutevole nistagmo.
Quando è indicato la neuroimaging,
la risonanza magnetica (MRI) pesata in diffusione con angiografia a risonanza
magnetica è attualmente considerato lo studio ottimale. Per gli ictus
emorragici, la tomografia computerizzata (TC) e la risonanza magnetica sono
entrambi ottimi studi. 31 Tuttavia, per l’ictus
ischemico, la RM è chiaramente superiore con una sensibilità dell'83% rispetto
al 26% della CT. 4 I medici non devono
quindi fare affidamento sulla TC per diagnosticare l’ infarto cerebellare.
CONCLUSIONE
L’Infarto cerebellare è
presente nel 3% dei pazienti che si presentano con le vertigini. 1 Di questi, solo il
10% non ha deficit neurologici focali. Le prove citate in questa revisione
suggeriscono che anche quando non ci sono deficit neurologici, la maggior parte
dei casi di infarto cerebellare si presenteranno sia con l'incapacità di
camminare senza sostegno o nistagmo che cambia direzione. 23 In un paziente con
una bassa probabilità a priori di ictus, il medico del pronto soccorso di
solito ha una buona giustificazione per non ricoverare un paziente che ha
vertigini isolate e un esame neurologico e neuro-otologico del tutto normal. Uno
studio di 15 casi di mancato infarto cerebellare ha dimostrato che in tutti e
15 mancava la documentazione di un esame neurologico normale e della marcia e dell'andatura. 3 La pronta valutazione
di un neurologo o di un otorinolaringoiatra è raccomandata nei pazienti che non
hanno ricevuto una diagnosi definitiva.
Conflitti di interesse: dall'accordo
articolo di presentazione Occidente JEM, tutti gli autori sono
tenuti a rivelare tutte le affiliazioni, fonti di finanziamento, e rapporti
finanziari o di gestione che possono essere percepite come potenziali fonti di
distorsione. Gli autori divulgati nessuno.
Supervisione curatore di sezione: Kurt R. Denninghoff,
MD
2. Kerber KA, Brown DL, Lisabeth LD, et
al. Stroke among patients with dizziness, vertigo, and imbalance in the
emergency department: a population-based
study. Stroke. 2006; 37 :2484–7. [ PMC free article ][ PubMed ]
3. Savitz SI, Caplan LR, Edlow JA. Pitfalls in
the diagnosis of cerebellar infarction. Acad Emerg
Med. 2007;14 :63–8. [ PubMed ]
4. Chalela JA, Kidwell CS, Nentwich LM, et
al. Magnetic resonance imaging and computed tomography in emergency
assessment of patients with suspected acute stroke: a prospective
comparison. Lancet. 2007; 369:293–8. [ PMC free article ] [ PubMed ]
5. Neuhauser HK. Epidemiology of
vertigo. Curr Opin Neurol. 2007; 20 :40–6. [ PubMed ]
7. Drachman DA, Hart CW. An approach to the
dizzy patient. Neurology. 1972; 22 :323–34. [ PubMed ]
8. Newman-Toker DE, Cannon LM, Stofferahn ME, et
al. Imprecision in patient reports of dizziness symptom quality: a
cross-sectional study conducted in an acute care setting. Mayo Clin Proc. 2007; 82:1329–40. [ PubMed ]
9. Neuhauser HK, von Brevern M, Radtke A, et
al. Epidemiology of vestibular vertigo: a neurotologic survey of the
general
population. Neurology. 2005; 65 :898–904. [ PubMed ]
10. Baloh RW. Differentiating between
peripheral and central cause of vertigo. Otolaryngol Head Neck
Surg. 1998; 119 :55–59. [ PubMed ]
12. Dix MR, Hallpike CS. The pathology,
symptomatology and diagnosis of certain common disorders of the vestibular
system. Proc R Soc Med. 1952; 45 :341–354. [ PMC free article ] [ PubMed ]
13. Viirre E, Purcell I, Baloh RW. The
Dix-Hallpike test and the canalith repositioning
maneuver.Laryngoscope. 2005; 115 :184–7. [ PubMed ]
14. Committee on Hearing and Equilibrium, American
Academy of Otolaryngology-Head and Neck Foundation, Inc Guidelines for the
diagnosis and evaluation of therapy in Meniere's disease. Otolaryngol Head
Neck Surg. 1995; 113 :181–5. [ PubMed ]
15. Son EJ, Bang JH, Kang JG. Anterior inferior
cerebellar artery infarction presenting with sudden hearing loss and
vertigo. Laryngoscope. 2007; 117 :556–8. [ PubMed ]
16. Lee H, Baloh RW. Sudden deafness in
vertebrobasilar ischemia: clinical features, vascular topographical patterns
and long-term outcome. J Neurol
Sci. 2005; 228 :99–104. [ PubMed ]
17. Kentala E. Characteristics of six otologic
diseases involving vertigo. Am J
Otol. 1996; 17 :883–92.[ PubMed ]
18. Brantberg K, Trees N, Baloh
RW. Migraine-associated vertigo. Acta
Otolaryngol. 2005; 125 :276–9.[ PubMed ]
19. Neuhauser H, Lempert T. Vertigo and dizziness
related to migraine: a diagnostic
challenge. Cephalalgia.2004; 24 :83–91. [ PubMed ]
20. von Brevern M, Zeise D, Neuhauser H, et
al. Acute migrainous vertigo: clinical and oculographic
findings. Brain. 2005; 128 :365–74. [ PubMed ]
21. Olesen J. Classification and diagnostic criteria
for headache disorders, cranial neuralgias and facial pain.Cephalalgia. 1988; 8 :9–96. [ PubMed ]
23. Lee H, Sohn SI, Cho YW, et al. Cerebellar
infarction presenting isolated vertigo: frequency and vascular topographical
patterns. Neurology. 2006; 67 :1178–83. [ PubMed ]
24. Halmagyi GM, Curthoys IS. A clinical sign
of canal paresis. Arch
Neurol. 1988; 45 :737–9. [ PubMed ]
25. Lewis RF, Carey JP. Abnormal eye movements
associated with unilateral loss of vestibular function. N Engl J
Med. 2006; 355 :e26. [ PubMed ]
26. Newman-Toker DE, Kattah JC, Alvernia JE, et
al. Normal head impulse test differentiates acute cerebellar strokes from
vestibular
neuritis. Neurology. 2008; 70 :2378–85. [ PubMed ]
27. Beynon GJ, Jani P, Baguley DM. A clinical
evaluation of head impulse testing. Clin
Otolaryngol. 1998;23 :117–22. [ PubMed ]
28. Harvey SA, Wood DJ. The oculocephalic
response in the evaluation of the dizzy
patient. Laryngoscope.1996; 106 :6–9. [ PubMed ]
29. Tohgi H, Takahashi S, Chiba K, et
al. Cerebellar infarction. Clinical and neuroimaging analysis in 293
patients. The Tohoku Cerebellar Infarction Study
Group. Stroke. 1993; 24 :1697–701. [ PubMed ]
30. Leigh RJ, Rucker JC. Nystagmus and related
ocular motility disorders. In: Miller NR, Newman NJ, editors. Walsh
and Hoyt's Clinical Neuro-Opthalmology. Baltimore, MD: Lippincott,
Williams & Wilkins;2004.
31. Kidwell CS, Chalela JA, Saver JL, et
al. Comparison of MRI and CT for detection of acute intracerebral
hemorrhage. JAMA. 2004; 292 :1823–30. [ PubMed ]
I disturbi che riguardano
il cervelletto, e le sue afferenze ed efferenze, hanno come conseguenza
alterazioni della velocità, dell'ampiezza e della forza dei movimenti. Dal
punto di vista anatomico il cervelletto è diviso in tre parti. L'archicerebello
o vestibolocerebello, comprende il lobo flocculonodulare ed è preposto al
mantenimento dell'equilibrio e dei movimenti coordinati occhi-testa-collo. È in
stretta connessione con i nuclei vestibolari. Il verme mediano
(paleocerebellum) coordina i movimenti del tronco e delle gambe: le lesioni a
questo livello comportano alterazioni della postura e dell'andatura. Gli
emisferi laterali, che costituiscono il neocerebellum, esercitano il controllo
sui movimenti balistici e finemente coordinati degli arti, in particolare di
quelli superiori. I segni delle malattie cerebellari sono riportati nella Tab.
1.
TABELLA 1. SEGNI DI MALATTIA
CEREBELLARE
Segno
Descrizione
Atassia
Barcollamento, andatura a base
allargata
Scomposizione del movimento
Incapacità di effettuare in
sequenza appropriata atti fini e coordinati
Disartria
Incapacità di articolare le parole
in modo appropriato, con farfugliamento e composizione inappropriata delle
frasi
Adiadococinesia
Incapacità di effettuare movimenti
rapidi e alternati
Dismetria
Incapacità di controllare
l'ampiezza del movimento
Ipotonia
Diminuzione del tono muscolare
Nistagmo
Oscillazione rapida e involontaria
dei globi oculari in direzione orizzontale, verticale o rotatoria con la
componente veloce massima verso la sede della lesione cerebellare
Parola scandita
Enunciazione lenta con tendenza a
esitare all'inizio della parola o della sillaba
Tremore
Movimento ritmico, alternante e
oscillatorio di un arto mentre questo si avvicina a un bersaglio (tremore
intenzionale) o della muscolatura prossimale mentre si tenta di mantenere una
postura o un peso (tremore di mantenimento)
LESIONI STRUTTURALI DEL
CERVELLETTO
Gli infarti, le emorragie
o i tumori, accrescendosi, possono essere causa di idrocefalo o di aumento
della pressione endocranica, con edema della papilla. Durante l'infanzia, la
parte mediana del cervelletto è la sede più frequente dei tumori primitivi
dell'encefalo (medulloblastoma, astrocitoma cistico). Deficit cerebellari
possono risultare da placche demielinizzanti nella sclerosi multipla (che
possono essere localizzate in qualunque parte della sostanza bianca
cerebellare), dalla malformazione di Chiari (discesa di tessuto cerebellare nel
canale cervicale) e dall'invaginazione basilare con platibasia (appiattimento
della base del cranio).
L'alcolismo e i problemi
nutrizionali, a esso collegati, possono dare luogo a una degenerazione del
verme e della parte anteriore del cervelletto con grave atassia dell'andatura.
Sindromi cerebellari possono anche essere secondarie a ipotiroidismo, a tossine
(monossido di carbonio, metalli pesanti, fenitoina), a iperpiressia, a traumi
cranici ripetuti. Meno frequentemente, nei bambini, disfunzioni cerebellari
reversibili possono essere secondarie a infezioni virali. Negli adulti, in
concomitanza con neoplasie maligne, possono manifestarsi, sebbene raramente,
gravi alterazioni estese a tutto il cervelletto.
DEGENERAZIONI
SPINO-CEREBELLARI
Gruppo di malattie
caratterizzate da atassia progressiva dovuta alla degenerazione del
cervelletto, del tronco, del midollo, dei nervi periferici e talvolta dei
gangli della base.
Molte di queste sindromi
sono ereditarie, mentre altre sono sporadiche. Le degenerazioni
spino-cerebellari possono essere classificate nelle seguenti ampie categorie:
le atassie a predominanza spinale, le atassie cerebellari, le degenerazioni
sistemiche multiple (v. Tab. 2). Non esiste terapia.
TABELLA
2.
PRINCIPALI
CARATTERISTICHE CLINICHE DI ALCUNE MALATTIE SPINOCEREBELLARI
L'atassia di Friedreich
rappresenta il quadro più tipico di atassia spinale. La trasmissione genetica è
di tipo autosomico recessivo; il gene responsabile è localizzato sul cromosoma
9. L'andatura incerta inizia tra i 5 e i 15 anni ed è seguita da atassia degli
arti superiori e disartria; le capacità mentali spesso diminuiscono. Il
tremore, anche se presente, è minimo. I pazienti sono areflessici e c'è una
perdita del tipo di sensibilità trasportato dalle fibre di largo calibro (senso
della vibrazione e della posizione); sono di frequente riscontro piede talo,
scoliosi e cardiomiopatia evolutiva. L'abetalipoprotidemia
(sindrome di Bassen-Kornzweig, deficienza di vitamina E) e la malattia di
Refsum (accumulo di acido fitanico ), hanno alcuni caratteri in comune con
l'atassia di Friedreich, rimanendo sconosciuta la causa metabolica di
quest'ultima.
L'atassia cerebellare
inizia in genere tra i 30 e i 50 anni, può essere di tipo sporadico o
ereditaria con carattere dominante. I reperti anatomopatologici sono limitati
al cervelletto e, talvolta, alle olive inferiori. Clinicamente, possono essere
evidenziati solo segni di disfunzione cerebellare.
Nell'atrofia
multisistemica (atrofia olivopontocerebellare), l'atassia
insorge nell'età media giovanile. Sintomi aggiuntivi comprendono la spasticità
in varie combinazioni con deficit extrapiramidali, sensitivi, del motoneurone
inferiore e del sistema autonomo. In alcuni gruppi familiari sono riscontrabili
l'atrofia ottica, la retinite pigmentosa, l'oftalmoplegia e la demenza. Tali
sindromi comprendono la sindrome dominante di Menzel (con deficit dei nervi
cranici e spasticità), la sindrome di Dejerine-Thomas, sporadica o recessiva,
in cui è prevalente il parkinsonismo, la degenerazione sistemica motoria delle
Azzorre (malattia di Machado-Joseph) e l'atassia cerebellare con alterazioni
neurovegetative (sindrome di Shy-Drager).
Alcuni
disturbi sistemici a patogenesi sconosciuta, quali l'atassia-telangiectasia
, producono atassia. Nel disturbo multisistemico mitocondriale,
l'atassia si manifesta in modi diversi associandosi a oftalmoplegia, blocco
cardiaco, miopatia. L'attività di molti enzimi della catena respiratoria
risulta diminuita, sono presenti delle delezioni nel DNA mitocondriale e alla
biopsia muscolare si evidenziano le caratteristiche fibre sfrangiate. L’inquadramento
nosografico delle malattie cerebellari degenerative è stato da sempre ed è
tuttora materia di discussione e di controversie. Per lungo tempo hanno
prevalso classificazioni di tipo neuropatologico (atassie spino-cerebellari,
olivo-ponto-cerebellari, atrofie cerebellari corticali). Molte affezioni sono
state a lungo citate con l’eponimo degli autori che le avevano descritte.
Negli ultimi anni sono comparse e sono sempre più utilizzate classificazioni
fondate su aspetti di ereditarietà o sporadicità e infine di genetica
molecolare che sembrano aver reso più semplice e razionale questo problema
classificativo. Sicuramente la quota relativa alle forme ereditarie è
numericamente nettamente prevalente su quella delle forme sporadiche e
comprende un vasto elenco di malattie fenotipicamente
te e genotipicamente diverse che peraltro sono di eccezionale riscontro nella
comune pratica clinica. In questo gruppo di “atassie ereditarie” prevalgono
quelle con contemporanea compromissione midollare (“eredo-atassie
spino-cerebellari”) che a loro volta si dividono, a seconda del modo di
trasmissione che le caratterizza, in due gruppi: atassie cerebellari
autosomiche dominanti (ADCA) ed autosomiche recessive (ARCA). La
prevalenza delle prime è stimata tra 0,8 e 5 casi per 100.000 mentre per
le seconde si stimano complessivamente circa 7 casi per 100.000, per la massima
parte dovuti alla atassia di Friedreich ed in minor misura alla
Atassia-teleangectasia di Louis-Bar. Per quanto riguarda il meccanismo
patogenetico che determina l’atassia, queste affezioni si possono attualmente
riunire in tre gruppi che pur avendo in comune un processo di progressiva
perdita neuronale, non condividono la natura di “degenerativa” nel senso
stretto del termine che verrebbe ad essere applicabile solo al terzo gruppo:
— atassie metaboliche a diversa età di esordio, secondarie a variazioni di geni
che regolano i livelli cerebrali e tissutali di metaboliti cerebrali che
possono divenire tossici ad anomale concentrazioni per il sistema cellulare
cerebellare;
—
atassie congenite, secondarie ad alterazioni di geni homeobox che
regolano lo sviluppo del complesso sistema cellulare cerebellare;
—
atassie degenerative, in relazione all’attivazione di geni che portano alla neuro
degenerazione ed all’apoptosi.
Infine, sia nell’ambito delle ADCA che in quello delle ARCA esistono numerose atassie
spino cerebellari geneticamente definite che vengono indicate utilizzando
l’acronimo SCA (spino-cerebellar-atrophy)seguito da un
numero arabo. Sinora ne sono state caratterizzate oltre 35 e la maggior
parte di esse appartengono alle ADCA ove sono ripartite in tre categorie
fenotipiche. Nel tipo I (caratterizzato da sintomi cerebellari e non
cerebellari) sono incluse le SCAI - SCA4, SCA8, SCAlO, SCA12 - SCA2,3, SCA25,
SCA27, SCA28, SCA32 e 5CA36; nel tipo Il (sindromi cerebellari ed anomalie
oculari a carico della macula) vi è la SCA7 e nel tipo ifi (sindromi
esclusivamente o prevalentemente cerebellari) sono inserite le SCA5, SCA6,
SCAli, SCA26, SCA3O e la SCA31.
La diagnosi di disturbi
cerebellari
Le principali
caratteristiche cliniche dei disturbi cerebellari sono incoordinazione,
squilibrio, e problemi con la stabilizzazione dei movimenti oculari. Ci sono
due distinguibili sindromi cerebellari - linea mediana e emisferica.
Sindromi cerebellari
Midline sono caratterizzate da uno squilibrio. Le persone sono instabili, non
sono in grado di stare in Romberg con gli occhi aperti o chiusi, e non sono in
grado di svolgere bene in tandem andatura. Linea mediana grave anomalia causa
"atassia trunkal" una sindrome in cui una persona è in grado di
sedersi sul loro letto senza stabilizzante se stessi. Alcune persone hanno
"titubation" o un movimento bobbing della testa o del tronco.
Disturbi cerebellari linea mediana anche spesso colpiscono i movimenti oculari.
Ci possono essere nistagmo, dismetria oculare e poveri inseguimento.
Sindromi cerebellari
emisferiche sono caratterizzate da incoordinazione degli arti. Ci possono
essere decomposizione del movimento, dismetria, e rimbalzo. Dysdiadochokinesis
è la performance irregolare dei movimenti alternati rapidi. Tremori Intention
possono essere presenti su un tentativo di toccare un oggetto. Un tremito
cinetica può essere presente in movimento. I test dito-to-naso e
tacco-to-ginocchio sono test classici della disfunzione cerebellare
emisferica. Mentre i riflessi possono essere depressi inizialmente con
sindromi cerebellari emisferiche, questo non si può contare. Discorso può
essere disartrico, scansione, o avere l'accento irregolare sillabe.
La diagnosi di
laboratorio di Malattie cerebellari
La diagnosi di un
disturbo cerebellare è generalmente costituito da un neurologo, e di solito è
semplice, a causa della elevata specificità dei segni sopra descritti.
ENG
o test sedia rotatoria possono mostrare segni specifici di un disturbo
cerebellare. In generale, si deve essere molto attenti nell'uso di questi
studi come audiologi che comunemente interpretano test ENG, generalmente non
hanno familiarità con disturbi centrali, e spesso semplicemente dire che il
paziente ha un "disturbo centrale vestibolare", piuttosto che
indicano che don ' t trovare qualcosa di sbagliato con le orecchie dei loro
pazienti. Di più su segni cerebellari nelle porzioni saccadici della ENG /
test sedia rotatoria si trovano qui.
Brain
Imaging
Brain imaging is always obtained in cerebellar disorders.L'imaging cerebrale si
ottiene sempre nei disturbi cerebellari. MRI
imaging, with the highest filed strength that is available, should be
undertaken. RMN, con la più alta forza depositato che è disponibile,
dovrebbe essere intrapreso. We do not recommend
"open MRI" testing in MRI's, or "low field". Si
consiglia di non test "risonanza magnetica aperta" a risonanza
magnetica, o "campo di basso". At the present
writing - -2008 -- we recommend 3T MRI testing, preferably with T1, T2,
diffusion, and Flair. Al momento di scrittura - -2008 - si consiglia il
test 3T MRI, preferibilmente con T1, T2, la diffusione, e Flair. T1 sagittal images must be obtained to diagnose the Chiari malformation . T1 immagini sagittali devono essere
ottenuti per diagnosticare la malformazione di Chiari.
In general, MRI
scanning often shows shrinkage of part or all of the cerebellum and/or
shrinkage of the brainstem.
In generale, la scansione MRI mostra spesso ritiro parziale o totale del
cervelletto e / o contrazione del tronco cerebrale. While
great progress has been made recently in the identification of genetic causes of
cerebellar atrophy , nevertheless the most common situation is for genetic testing
(if available) to be negative.
Sebbene siano stati compiuti grandi progressi recentemente nel identificazione
di cause genetiche di atrofia
cerebellare,
tuttavia la situazione più comune è per il test diagnostico (se disponibile)
per essere negativo. This means that genetic testing
is frequently unhelpful, and the role of cerebellar genetic testing should not
be overemphasized. Ciò significa che i test genetici è spesso inutile, e
il ruolo dei test genetici cerebellare non deve essere sottovalutata. Some general references about radiological diagnosis are
Huang, Tuason et al. Alcuni riferimenti generali sulla diagnosi
radiologica sono Huang, Tuason et al. 1993;
1993; Wullner, Klockgether et al. Wüllner,
Klockgether et al. 1993. 1993.
Because genetic testing
is usually negative in progressive cerebellar disorders, the
"diagnosis" of cerebellar disorders basically means separation of
patients into overlapping groups -- genetically identified degenerations,
progressive hereditary conditions without an identified gene, and undiagnosed causes
of cerebellar symptoms.
Poiché i test genetici è generalmente negativo nei disturbi cerebellari
progressive, la "diagnosi" di disturbi cerebellari in pratica
significa la separazione dei pazienti in gruppi sovrapposti - degenerazioni
geneticamente identificati, condizioni ereditarie progressive senza un gene
identificato, e le cause non diagnosticati di sintomi cerebellari. Generally the "undiagnosed" group is the largest
one. Generalmente il gruppo "non diagnosticata" è il più
grande.
Broadly speaking, there are many disorders that cause shrinkage
of the cerebellum -- such as hereditary degenerations or toxins.
In generale, ci sono molti disturbi che causano il restringimento del
cervelletto - come degenerazioni ereditarie o tossine. There are very few disorders that cause shrinkage of the brainstem -
-these are mainly hereditary degenerations. Ci sono pochissimi i
disturbi che causano il restringimento del tronco encefalico - -Questi sono
degenerazioni principalmente ereditarie. Severe
cerebellar symptoms with a normal MRI scan suggest a paraneoplastic cerebellar problem . Gravi sintomi cerebellari con normale
risonanza magnetica suggeriscono un problema
cerebellare paraneoplastica.
In questa sede, tenendo conto delle finalità di questa trattazione, ci si limiterà
a riportare aspetti di interesse otoneurologico riguardanti in generale questo
gruppo di malattie passando successivamente a descrivere gli aspetti clinici
essenziali della sindrome più frequente di ciascuno dei due gruppi di atassie
ereditarie e cioè dell’atassia spinocerebellare di Friedreich per le ARCA e
della SCA6 per le ADCA, Le forme caratterizzate da degenerazione isolata del
cervelletto sono di fatto piuttosto rare poiché nella maggior parte dei casi al
danno cerebellare coesistono segni e sintomi dì interessamento di altre
strutture anatomicamente e/o funzionalmente collegate col cervelletto come il
tronco encefalico ed il midollo spinale. Da ciò consegue che anche la
fenomenologia vestibolare risulta generalmente assai complessa esprimendo, non
solo lo stato di sofferenza delle strutture cerebellari interessate dalla
lesione, ma anche quello dei distretti nervosi adiacenti come i nuclei
vestibolari troncoencefalici, le connessioni cerebello-ponto-mesencefaliche e
quelle cerebello-vestibolospinali. Sul piano sintomatologico, nei soggetti con
sofferenza cerebellare isolata, la vertigine è solitamente un elemento del
tutto secondario e spesso manca del tutto con l’eccezione delle localizzazioni
vermiane mentre è frequente quando si associano lesioni a carico delle strutture
vestibolari bulbo-pontine. Non è rara l’oscillopsia dovuta al deficit della
soppressione cerebellare del VOR.
Nelle
lesioni cerebellari degenerative a decorso lento si può avere una deviazione
obliqua (skew deviation) dei bulbi oculari ed anche il nistagmo
spontaneo può mancare ed essere sostuituito da un nistagmo da direzione dello
sguardo o da un nistagmo verticale spesso con pattern irregolare della
componente rapida.
Nelle forme unilaterali il nistagmo è diretto verso il focolaio, salvo che in
certe patologie del corpo restiforme nelle quali può essere diretto verso il
lato opposto. Nelle lesioni della linea mediana può frequentemente osservarsi
un nistagmo verticale, alterazioni del nistagmo otto- cinetico, anomalie a
carico delle saccadi (dismetria), del pursuit e della stabilità oculare durante
la fissazione e sono inoltre decisamente alterate le. prove di postura statica
e dinamica (Vaii de Warrenburg et al., 2005) e quelle per le asimmetrie
segmentarie toniche.
Atassia
Telangiectasia (AT) o sindrome di Louis-Bar,
L’atassia-teleangectasia o sindrome
di Louis-Bar (A-T) è l'associazione tra
un'immunodeficienza combinata grave (che interessa in particolare la risposta
immuno-umorale) e un'atassia cerebellare progressiva; è una malattia genetica a
trasmissione autosomica recessiva multisistemica caratterizzata da: atassia
cerebellare degenerazione del cervelletto teleangectasie oculo-cutanee da immunodeficienza.Ataxia-telangiectasia is an autosomal recessive
multisystem disease.
È caratterizzata da segni
neurologici, teleangectasie, suscettibilità alle infezioni e rischio elevato di
sviluppare un tumore. Other features include a
variable immunodeficiency, endocrinopathy, respiratory failure, a high risk for
malignancy, increased serum alpha-fetoprotein levels, radiosensitivity, and
chromosomal instability. Altre caratteristiche includono una
immunodeficienza variabile, endocrinopatia, insufficienza respiratoria, un alto
rischio di malignità, i livelli di alfa-fetoproteina aumentato siero,
radiosensibilità e instabilità cromosomica.
Most persons with AT
are unsteady when they first start to walk, and become wheelchair bound at
approximately the age of 10. Oculomotor apraxia, with characteristic head
thrusts, are common in AT.
La maggior parte delle persone con AT sono instabili quando iniziano a
camminare, e diventano sedia a rotelle a circa all'età di 10. Oculomotor
aprassia, con caratteristiche spinte testa, sono comuni in AT. In most persons, death occurs in the third decade of life
due to malignacy or respiratory failure. Nella maggior parte delle
persone, la morte avviene nella terza decade di vita a causa di insufficienza
respiratoria.
There are rare variants
of AT in whom features are milder and survival is longer. Ci sono varianti rare di AT nei
quali caratteristiche sono più miti e la sopravvivenza è più lungo. (MMM Verhagen et al, 2009). (MMM Verhagen et al,
2009). In the variant form, the ocular
telangiectasia are milder or absent, and basal ganglia findings predominate over
cerebellar signs. Nella forma variante, la telangiectasia oculare sono
più lievi o assenti, ei risultati dei gangli della base predominano su segni
cerebellari.
Storia
La prima descrizione di questa sindrome risale al 1926,
quando due ricercatori cecoslovacchi, Ladislav Syllaba e Kamil Henner,
riportarono nel primo numero della Révue Neurologique tre casi
di quella che chiamarono atetosi doppia congenita. Nel 1941,
la giovane neurologa BelgaDenise Louis-Bar ne fornì una
descrizione più completa e, in seguito al suo lavoro, la sindrome prese il suo
nome. Determinanti sono stati gli studi condotti alla fine degli anni cinquanta da Elena Boder e Robert P.
Sedgwick, che raccolsero numerosi casi e introdussero per primi la
denominazione atassia-teleangectasia.
L'anomalia genetica che determina
l'atassia-teleangectasia è stata riconosciuta a livello del gene detto
ATM (ataxia-telangiectasia mutated),che
è stato clonato nel 1995. [2] ATM si trova sul cromosoma umano 11 (11q22.3) e
si compone di 69 esoni sparsi in 150kb di DNA genomico. [ 51 mappato nella
regione q22-23 del cromosoma11.L'A-T
è una malattia autosomica recessiva causata dalle mutazioni inattivanti del
gene ATM (11q22.3), che è espresso ubiquitariamente e codifica per una chinasi
proteica che svolge un ruolo nel controllo della riparazione delle rotture a
doppio filamento (DSB) nel DNA, in particolare nelle cellule di Purkinje del
cervelletto e nelle cellule cerebrali, nell'endotelio della congiuntiva e della
cute.Ogni genitore è un portatore,
significa che hanno una copia normale del gene AT (ATM) e da una copia che è
mutato. AT si verifica se un bambino eredita il mutato AT gene da ciascun
genitore, così in una famiglia con due genitori portatori, c'è 1 probabilità su
4 che un bambino nato da genitori avranno il disturbo. La diagnosi prenatale (e
individuazione del portatore) può essere effettuati in famiglie se sono stati
individuati gli errori (mutazione) in due geni ATM di un figlio affetto. Il
processo di ottenere questo fatto può essere complicato e, in quanto richiede
tempo, dovrebbe essere organizzato prima concezione.Cerchi mutazioni nel gene ATM
di una persona estranea (ad esempio, il coniuge di un noto vettore AT) presenta
sfide significative. I geni hanno spesso varianti ortografiche (polimorfismi)
che non influenzano la funzione. In un gene grande come bancomat, tali varianti
ortografiche sono probabili e medici non possono sempre prevedere se una
variante specifica sarà o non causare la malattia. La consulenza genetica può
aiutare i familiari di un paziente AT capire che cosa può o non può essere
provata, e di come i risultati del test dovrebbero essere interpretati.
Portatori
di AT, come i genitori di una persona con AT, hanno una copia mutata del gene
ATM e una copia normale. Sono generalmente in buona salute, ma vi è un aumento
del rischio di cancro al seno nelle donne. Questo dato è stato confermato in
una varietà di modi diversi, ed è l'oggetto di ricerca corrente. Sorveglianza
standard (compresi seno auto-esami mensili e mammografia al consueto orario per
l'età) è consigliato, a meno che ulteriori prove devono essere indicati perché
l'individuo ha altri fattori di rischio (per esempio, la storia familiare di
cancro al seno). La malattia A-T-simile è una variante rara di A-T dovuta
all'inattivazione del gene MRE11 (11q21), che codifica per una proteina
coinvolta nella riparazione delle DSB. È problematica la diagnosi clinica
precoce, ma l'aumento pressoché costante nel siero dei livelli di
alfa-fetoproteina (AFP) e l'analisi citogenetica possono contribuire a
confermare la diagnosi (traslocazioni 7;14). A volte è necessaria la diagnosi
molecolare
L'incidenza
dell'atassia-teleangectasia nella popolazione è stimata da 1 su 40.000 fino a 1
su 100.000 nati vivi [Charles et al., 2000][3] [57].
In
Italia i casi conosciuti a tutt'oggi (2012) sono circa 50 quindi l'incidenza è
molto inferiore.
Per
l’AT non esiste allo stato attuale una terapia risolutiva, ma una diagnosi
precoce potrebbe consentire di evitare le complicanze infettive e neoplasiche;
inoltre permetterebbe alle famiglie una consulenza genetica, per prevenire
eventuali rischi in gravidanze successive.
I ricercatori californiani hanno
riscontrato la possibilità di diagnosticare la patologia durante lo screening
neonatale per la SCID (immunodeficienza combinata grave), patologia caratterizzata
dal mancato sviluppo dei linfociti T e dalla grave compromissione del sistema
immunitario, incapace di difendersi dagli agenti infettivi. Per diagnosticare
la SCID si usa una particolare metodologia, in grado di rilevare la presenza di
un particolare recettore dei linfociti T chiamato TREC. Questo esame, eseguito
in diversi stati americani, permette di identificare la carenza di linfociti T
tipica della SCID, che però può essere anche una caratteristica dell’ Atassia
Telangiectasia.
Nel caso di linfocitopenia, se la
diagnosi di SCID non può essere confermata, è necessario eseguire il
sequenziamento dell’esoma, grazie al quale è possibile identificare la
mutazione del gene ATM, causa di atassia telangiectasia.
Grazie a questa ricerca sono stati
individuati diversi casi di AT che non presentavano i sintomi clinici tipici.
E’ stato possibile inoltre effettuare una ricerca retrospettiva sui campioni di
sangue secco depositati molti anni prima, grazie ai quali sono stati
diagnosticati alcuni casi atipici di AT in ragazzi adolescenti
Descrizione clinica
Atassia
L'atassia compare in genere nella
prima infanzia e si rende evidente nel giro di pochi anni con progressivi
disturbi della postura e dell'andatura, con incapacità di compiere movimenti
fini (utilizzo delle posate, della matita, di piccoli oggetti) e con aprassia oculomotoria, cioè con
l'incapacità di seguire gli oggetti in movimento attraverso il campo visivo: il
bambino ruota la testa anziché muovere gli occhi. Compaiono successivamente
disturbi del linguaggio e diminuzione della capacità muscolare, soprattutto a
carico degli arti inferiori. In età adolescenziale, in genere tutti i pazienti
necessitano di un consistente aiuto per vestirsi, mangiare e provvedere
all'igiene personale.
Le capacità intellettive non vengono
compromesse, ma è difficile valutare l'intelligenza di un soggetto affetto da
questa sindrome per la lentezza delle risposte.
Teleangectasia
Le teleangectasie sono dilatazioni
dei piccoli vasi che si manifestano dapprima a livello delle congiuntive, poi si diffondono su tutta la
cute, soprattutto sulle orecchie, sopra il ponte del naso,
nelle pieghe del gomito e del ginocchio. Occasionalmente possono essere
presenti alla nascita, ma in genere si sviluppano dopo la prima decade di età.
Di solito non costituiscono un problema medico in quanto non sanguinano e non
danno prurito. Rispetto alle teleangectasie che
si ritrovano nell'occhio di persone più anziane, queste sono costanti nel tempo
e non peggiorano o migliorano col variare della temperatura corporea, dei fattori
emozionali e dello stato di sanguificazione dell'individuo.
Immunodeficienza
L'immunodeficienza esordisce con
infezioni respiratorie (in particolare a livello polmonare) che col tempo
recidivano fino a portare allo sviluppo di bronchiectasie. Spesso i pazienti soffrono
di riniti e rinosinusiti: la ridotta capacità di combattere
le infezioni riguarda prevalentemente le malattie causate da batteri piuttosto che quelle da virus.
Quasi mai, tuttavia, si rileva un deficit di gammaglobuline.
Conseguenze importanti del
malfunzionamento del sistema
immunitario sono
l'aumentata suscettibilità alle radiazioni
ionizzanti, e
ai tumori. L'esposizione a raggi X in questi soggetti va pertanto
limitata a situazioni di assoluta necessità, per la loro incapacità di riparare
i danni del DNA. Le neoplasie più frequenti nei
soggetti affetti da atassia-teleangectasia sono i linfomi e leleucemie
linfatiche, ma nei
soggetti che sopravvivono più a lungo si riscontrano anche carcinomi.
Treatment:Trattamento:
Recently it has been
reported that 4-AP may be helpful. La presa in carico è sintomatica e
comprende la fisioterapia, la terapia del linguaggio e il trattamento delle
infezioni e delle complicazioni polmonari. Dato che le cellule dei pazienti
affetti da A-T mostrano suscettibilità ai raggi X, la radioterapia, associata
ad alcune forme di chemioterapia, deve essere usata con cautela. I bambini
affetti spesso necessitano della sedia a rotelle all'età di 10-11 anni. La
prognosi è grave, per la possibilità di insorgenza di infezioni respiratorie,
per la neurodegenerazione, l'invecchiamento cutaneo-mucoso precoce e il rischio
di sviluppare tumori (il 35% dei pazienti sviluppa un cancro prima dei 20
anni).Recentemente è stato riportato che 4-AP può essere utile.
Atassia ed altri problemi neurologici
Non esiste un
trattamento noto per rallentare o arrestare la progressione dei problemi
neurologici. Il trattamento di AT è sintomatico e di
supporto. Fisiche, terapie ed esercizi professionali e vocali possono
aiutare a mantenere la funzione, ma non rallentare il decorso della neurodegenerazione. Esercizi
terapeutici non dovrebbero essere usati al punto di fatica e non dovrebbero
interferire con le attività della vita quotidiana. Alcuni farmaci
anti-Parkinson e antiepilettici forse utili nella gestione dei sintomi, ma
dovrebbe essere prescritto in consultazione con un neurologo.
Problemi immunitari
Tutti gli individui
con AT dovrebbero avere almeno una valutazione completa immunologica che misura
il numero e il tipo di linfociti nel sangue (linfociti T e linfociti B), i
livelli di immunoglobuline sieriche (IgG, IgA e IgM) e risposta anticorpale a T-dipendente
(ad esempio, il tetano, Haemophilus influenzae b) e T-indipendenti (23-valente
pneumococcico polisaccaridico) vaccini. Per la maggior parte, il modello
di immunodeficienza visto in un paziente AT primi anni di vita (per cinque
anni) sarà lo stesso modello visto per tutta la durata di
quell'individuo. Pertanto, le prove non devono essere ripetute, a meno che
individuo sviluppa più problemi con l'infezione. I problemi con l'immunità
a volte possono essere superate immunizzazione. I vaccini contro patogeni
respiratori batteri comuni come Haemophilus influenzae, pneumococchi e virus
dell'influenza (la "influenza") sono disponibili in commercio e
spesso aiutano a stimolare risposte anticorpali, anche in soggetti con bassi
livelli di immunoglobuline. Se i vaccini non funzionano e il paziente
continua ad avere problemi con infezioni, la terapia gammaglobuline (IV o
infusioni sottocutanee di anticorpi raccolti da soggetti normali) può essere di
beneficio. Un piccolo numero di persone con AT sviluppano un'anomalia in
cui uno o più tipi di immunoglobuline sono aumentati di gran lunga superiori a
quelle normali. In alcuni casi, i livelli di immunoglobuline possono
essere aumentati tanto che il sangue diventa denso e non scorre
correttamente. La terapia per questo problema deve essere adattata per
l'anomalia specifica trovata e la sua gravità.
Se la suscettibilità
di un singolo paziente per infezione aumenta, è importante rivalutare è
verificato funzione immunitaria in caso di deterioramento e una nuova terapia viene
indicato. Se le infezioni si verificano nel polmone, è importante anche
per studiare la possibilità di rondine disfunzionale con l'aspirazione nei
polmoni (vedi sopra sezioni sotto
Sintomi:
Alimentazione, Deglutizione e Nutrizione.
La maggior parte delle
persone con AT hanno un basso numero di linfociti nel sangue. Questo
problema sembra essere relativamente stabile con l'età, ma un numero rara di
gente hanno progressivamente decrescenti conta dei linfociti che
invecchiano. Nella popolazione generale, molto bassa conta di linfociti
sono associati ad un aumento del rischio di infezione. Tali individui
sviluppano complicanze da vaccini virali vivi (morbillo, parotite, rosolia e
varicella), infezioni virali croniche o gravi, infezioni lievito della pelle e
della vagina, e infezioni opportunistiche (come ad esempio la polmonite
polmonite). Anche se conta dei linfociti sono spesso a partire in persone
con AT, raramente hanno problemi con infezioni opportunistiche. (L'unica
eccezione a questa regola è che i problemi con verruche cronici o ricorrenti
sono comuni.) Il numero e la funzione dei linfociti T devono essere rivalutati
se una persona con AT è trattata con farmaci corticosteroidi quali prednisone
per più di un paio settimane o è trattato con la chemioterapia per il
cancro. Se conta dei linfociti sono bassi in persone che prendono questi
tipi di farmaci, si consiglia l'uso di profilassi antibiotica per prevenire le
infezioni opportunistiche.
Se le prove dimostrano
notevoli alterazioni del sistema immunitario, uno specialista in malattie
infettive immunodeficienza o sarà in grado di discutere le varie opzioni di
trattamento. Assenza di immunoglobuline o anticorpi risposte al vaccino
può essere trattata con sostituzione gamma globulina infusi, o può essere gestito
con antibiotici profilattici e minimizzata l'esposizione alle
infezioni. Se la funzione di anticorpi è normale, tutte le vaccinazioni
infantili di routine, tra cui vaccini vivi virali (morbillo, parotite, rosolia
e varicella) dovrebbe essere data. Inoltre, diversi vaccini
"speciali" (ovvero, la licenza ma non di routine per i bambini
altrimenti sani e giovani adulti) devono essere somministrate per ridurre il
rischio che un paziente AT svilupperà infezioni polmonari. Il paziente e
tutti i membri della famiglia devono ricevere la influenza (influenza) di
vaccino ogni caduta. Le persone con AT che hanno meno di due anni di età
devono ricevere tre (3) dosi di un vaccino pneumococcico coniugato (Prevnar)
data a intervalli di due mesi. Persone di età superiore a due anni che non
sono stati precedentemente immunizzati con Prevnar dovrebbero ricevere due (2)
dosi di Prevnar. Almeno 6 mesi dopo l'ultima Prevnar è stato dato e dopo
che il bambino è di almeno due anni, il 23-valente vaccino pneumococcico dovrebbero
essere somministrati. L'immunizzazione con il 23-valente vaccino
pneumococcico deve essere ripetuto circa ogni cinque anni dopo la prima dose.
Nelle persone con AT
che hanno bassi livelli di IgA, ulteriori test deve essere eseguito per
determinare se il livello di IgA è basso o completamente assente. Se
assente, c'è un leggero aumento del rischio di una reazione
trasfusionale. Bracciali "Medical Alert" non sono necessari, ma
la famiglia e medico primario devono essere consapevoli che se non c'è un intervento
chirurgico elettivo necessità di trasfusione di globuli rossi, le cellule
devono essere lavati per diminuire il rischio di una reazione allergica.
Le persone con AT
hanno anche un rischio maggiore di sviluppare malattie infiammatorie autoimmuni
o croniche. Questo rischio è probabilmente un effetto secondario di loro
dell'immunodeficienza e non un effetto diretto della mancanza di proteina
ATM. Gli esempi più comuni di tali disturbi in AT comprendono
trombocitopenia immune (ITP), varie forme di artrite, e della vitiligine.
Malattia polmonare
Infezioni del seno e
polmonari ricorrenti possono portare allo sviluppo di malattie polmonari
croniche. [12] Tali infezioni
dovrebbero essere trattati con antibiotici appropriati per prevenire e limitare
il danno polmonare. La somministrazione di antibiotici dovrebbe essere
considerato quando i bambini e gli adulti hanno sintomi respiratori prolungati (superiori
a 7 giorni), anche a seguito di quello che si presumeva essere stata una
infezione virale. Per aiutare a prevenire malattie respiratorie da
patogeni respiratori comuni, la vaccinazione influenzale annuale devono essere
somministrati vaccini contro lo pneumococco e devono essere somministrati al
momento opportuno. Il trattamento antibiotico dovrebbe anche essere presa
in considerazione nei bambini con tosse cronica che sono produttivi di muco,
coloro che non rispondono alle aggressive tecniche di liquidazione polmonari e
nei bambini con secrezioni muco-purulenta dai seni o al torace. Una tosse
umida può anche essere associata con l'aspirazione cronica che deve essere
escluso attraverso appropriati studi diagnostici, ma l'aspirazione e le infezioni
respiratorie non sono necessariamente escludono a vicenda. Nei bambini e
negli adulti con bronchiectasie, terapia antibiotica cronica dovrebbe essere
considerata per rallentare la progressione della malattia polmonare cronica.
Coltura dei seni può
essere necessario per dirigere la terapia antibiotica. Questo può essere
fatto da un otorinolaringoiatriche (ENT) specialista. Inoltre,
broncoscopia diagnostica può essere necessario nelle persone che hanno
polmoniti ricorrenti, in particolare quelli che non rispondono o rispondono in
modo incompleto a un ciclo di antibiotici.
Liquidazione delle
secrezioni bronchiali è essenziale per una buona salute polmonare e può aiutare
a limitare lesioni provocate da infezioni polmonari acute e croniche. I
bambini e gli adulti con aumento delle secrezioni bronchiali possono trarre
beneficio dalla terapia del torace di routine con il metodo manuale, un
dispositivo a cappella o un giubbotto fisioterapia respiratoria.Petto
fisioterapia può aiutare a portare mucose dall'albero bronchiale più bassa,
tuttavia una tosse adeguata è necessaria per rimuovere le
secrezioni. Nelle persone che sono diminuiti di riserva del polmone e una
tosse debole, l'uso di un insufflatore-Essufflatore (tosse-assistita)
dispositivo può essere utile come terapia di mantenimento o durante le malattie
respiratorie acute per aiutare a rimuovere le secrezioni bronchiali dalle vie
aeree superiori. La valutazione di uno specialista Pneumologia però, deve
prima essere fatto per valutare correttamente l'idoneità del paziente.
I bambini e gli adulti
con tosse cronica secca, aumento del lavoro respiratorio (frequenza
respiratoria veloce, respiro corto a riposo o con le attività) e l'assenza di
un processo infettivo per spiegare sintomi respiratori dovrebbero essere valutati
per malattia polmonare interstiziale o un altro processo
intrapolmonare. Valutazione da uno pneumologo e una TAC del torace
dovrebbe essere considerato in soggetti con sintomi di malattia polmonare
interstiziale o ad escludere altri processi polmonari non infettive. Le
persone con diagnosi di malattia polmonare interstiziale possono beneficiare di
steroidi sistemici.
Nutrire, la deglutizione e la nutrizione
Assunzione orale può
essere aiutata da insegnare persone con AT come bere, masticare e deglutire in
modo più sicuro. La proprietà di trattamenti per problemi di deglutizione
dovrebbe essere determinata a seguito della valutazione da parte di un esperto
nel campo della patologia del linguaggio. Dietologi possono aiutare i
problemi di nutrizione ossequio raccomandando modifiche dietetici, come gli
alimenti ad alto contenuto calorico o integratori alimentari.
Una alimentazione
(gastrostomia) tubo è consigliato quando una delle seguenti si
verificano: [56]
·Un
bambino non può mangiare abbastanza per crescere o una persona di qualsiasi
età, non può mangiare abbastanza per mantenere il peso;
·L'aspirazione
è problematica;
·I
pasti sono stressanti o troppo lungo, interferendo con le altre attività.
Tubi di alimentazione
possono ridurre il rischio di aspirazione per consentire alle persone di
evitare liquidi o alimenti che sono difficili da digerire e fornire adeguate
calorie senza l'impegno stress e il tempo di pasti prolungati. Tubi
gastrostomia non impediscono alle persone di mangiare per bocca. Una volta
che un tubo è a posto, l'obiettivo generale dovrebbe essere quello di mantenere
il peso al percentile 10-25th.
Istruzione e socializzazione
La maggior parte dei
bambini affetti da AT hanno difficoltà a scuola a causa di un ritardo nei tempi
di risposta per visive, verbali o altri segnali, slurred discorso e
tranquilla (disartria),anomalie del controllo oculare (aprassia oculomotoria), e
compromissione del controllo motorio. Nonostante questi problemi, i
bambini con AT spesso godono scuola se possono essere fatti strutture adeguate
alla loro disabilità. La decisione circa la necessità di classi speciali
di educazione o di un aiuto supplementare nelle classi normali è fortemente
influenzata dalle risorse locali disponibili. Le decisioni circa il
corretto posizionamento educativo devono essere rivisitate tutte le volte che
le circostanze lo richiedano. Nonostante le loro numerose menomazioni
neurologiche, la maggior parte degli individui con AT sono molto socialmente
consapevoli e socialmente qualificati, e quindi beneficiare di relazioni con i
coetanei sostenute sviluppate a scuola. Alcuni individui sono in grado di
funzionare abbastanza bene nonostante le loro disabilità e pochi si sono
laureati da community colleges.
Molti dei problemi
riscontrati potranno beneficiare di una particolare attenzione, in quanto i
problemi sono spesso legati più a problemi "di ingresso e di uscita"
che alla perdita di valore intellettuale. Problemi con il controllo dei
movimenti degli occhi rendono difficile per le persone con AT di leggere, ma
comprendere più pienamente il significato e sfumature di testo che viene letto
a loro. I ritardi nella iniziazione parola e la mancanza di espressione
facciale far sembrare che non conoscono le risposte alle
domande. Riduzione dello sforzo di qualificati necessari per rispondere
alle domande, e un aumento del tempo disponibile per rispondere, è spesso
ricompensato da vera realizzazione. E 'importante riconoscere che la
disabilità intellettuale non è regolarmente una parte del quadro clinico di AT,
anche se il rendimento scolastico può essere ottimale a causa delle molte difficoltà
nella lettura, scrittura, e la parola. I bambini con AT sono spesso molto
consapevoli del loro aspetto, e si sforzano di apparire normale ai loro
coetanei e insegnanti. La vita all'interno del corpo atassica può essere
faticoso. Lo sforzo maggiore necessaria per mantenere le apparenze e
aumento di energia consumate nei movimenti tono ed extra anormali
contribuiscono alla fatica fisica e mentale. Di conseguenza, per alcuni un
giorno di scuola accorciato produce benefici reali.
Raccomandazioni
generali:
·Tutti
i bambini con AT bisogno di un'attenzione particolare alle barriere che
sperimentano a scuola. Negli Stati Uniti, questo prende la forma di un IEP
formale (programma educativo individualizzato).
·I
bambini con AT tendono ad essere ottimi risolutori di problemi. Il loro
coinvolgimento nella modalità di esecuzione migliore compiti dovrebbe essere
incoraggiata.
·Logopedisti
possono facilitare le capacità di comunicazione che consentono ai soggetti con
AT per ottenere i loro messaggi attraverso (usando le parole chiave contro
frasi complete) e insegnare le strategie per ridurre la frustrazione associata
con il tempo incremento necessario per rispondere alle domande (ad esempio, in
possesso di una mano e altri sulla necessità di concedere più tempo per le
risposte). Raramente utile sono le terapie discorso tradizionali che si
concentrano sulla produzione di suoni e rafforzamento dei muscoli labbra e
lingua specifici.
·Aiutanti
in aula può essere opportuno, in particolare per aiutare con incisione, il
trasporto attraverso la scuola, i pasti e in bagno. L'impatto di un
aiutante in relazioni con i coetanei deve essere attentamente monitorato.
·La
terapia fisica è utile per mantenere la forza e la salute cardiovascolare in
generale. La terapia a cavallo ed esercizi in piscina sono spesso ben
tollerati e divertente per le persone con AT. Tuttavia, nessuna quantità
di pratica rallenta la degenerazione cerebellare o migliorare la funzione
neurologica. Esercizio al punto di esaurimento dovrebbe essere evitato.
·Audizione
è normale per tutta la vita. Libri su nastro può essere un utile
complemento per materiali scolastici tradizionali.
·L'uso
precoce dei computer (in età prescolare) con il software di completamento delle
parole dovrebbe essere incoraggiata.
·Praticare
coordinamento (ad esempio, trave di equilibrio o esercizi di scrittura corsivi)
non è disponibile.
·La
terapia occupazionale è utile per la gestione delle abilità di vita quotidiana.
·Lasciare
il tempo di riposo, giorni più brevi, orario delle lezioni ridotto, ridotto i
compiti a casa, test modificati se necessario.
·Come
tutti i bambini, quelli con AT bisogno di avere obiettivi di sperimentare la
soddisfazione di fare progressi.
·Le
interazioni sociali con i coetanei sono importanti e dovrebbero essere presi in
considerazione per il posizionamento di classe. Per tutti relazioni con i
coetanei a lungo termine può essere la parte più gratificante della
vita; per quelli con AT stabilire queste connessioni in anni di scuola può
essere utile.
Cliniche e
supporto
Gli Stati Uniti, Regno
Unito, Australia, Israele, Paesi Bassi, Germania, Polonia, Norvegia e Giappone,
abbiamo cliniche specializzate per i pazienti con AT. Queste cliniche
ospitano squadre mediche multidisciplinari, tra cui neurologi, immunologi,
pneumologi e terapisti, in grado di affrontare le molteplici sfaccettature di
questa malattia.
Prognosi
L'aspettativa di vita
delle persone con AT è molto variabile. La media è di circa 25 anni, ma
continua a migliorare con i progressi della cura. I due più comuni cause
di morte sono malattie polmonari croniche (circa un terzo dei casi) e il cancro
(circa un terzo dei casi).
Direzioni di
ricerca
Un open-label di fase II della sperimentazione clinica studiando l'uso
di globuli rossi (eritrociti) carichi di desametasone sodio fosfato scoperto
che questo trattamento ha migliorato i sintomi e sembrava essere ben
tollerato. [58] Il trattamento
utilizza un sistema di attuazione dei farmaci utilizzando propri globuli rossi
del paziente come veicolo di consegna per il farmaco. [59] Dati gli altri
deficit immunologici presenti in individui con AT, permane la necessità di
valutare il potenziale terapeutico di steroidi ulteriormente, soprattutto per
quanto riguarda la durata di qualsiasi beneficio e la sua sicurezza a lungo
termine.
References: Riferimenti:
Bibliografia
^ Charles R. Scriver,
Arthur L. Beaudet, The Metabolic and Molecular Bases of Inherited
Disease, New York, McGraw-Hill, 2000, ISBN978-0-07-913035-8.
Cuneo-A;
Bigoni-R; Rigolin-GM; Roberti-MG; Milani-R; Bardi-A; Minotto-C; Agostini-P;
De-Angeli-C; Narducci-MG; Sabbioni-S; Russo-G; Negrini-M; Castoldi-G.Acquired
chromosome 11q deletion involving the ataxia telangiectasia locus in B-cell
non-Hodgkin's lymphoma: Correlation with clinicobiologic features; JOURNAL-OF-CLINICAL-ONCOLOGY.
JUL 2000; 18 (13) : 2607-2614
Kappenhagen-N;
Royer-N; Scheuplein-M; Schubert-R; Zielen-S. Characterisation of lymphocyte
subset in patients with ataxia teleangiectasia; MONATSSCHRIFT-KINDERHEILKUNDE.
OCT 1999; 147 (10) : 927-928,929-931
Kleier-S;
Herrmann-M; Wittwer-B; Varon-R; Reis-A; Horst-J .Clinical presentation and
mutation identification in the NBS1 gene in a boy with Nijmegen breakage
syndrome; CLINICAL-GENETICS. MAY 2000; 57 (5) : 384-387
Seidemann-K;
Henze-G; Beck-JD; Sauerbrey-A; Kuhl-J; Mann-G; Reiter-A.Non-Hodgkin's lymphoma
in pediatric patients with chromosomal breakage syndromes (AT and NBS):
Experience from the BFM trials; ANNALS-OF-ONCOLOGY. 2000; 11 Suppl. 1 : 141-145
Seidemann-K;
Tiemann-M; Henze-G; Sauerbrey-A; Muller-S; Reiter-A.Therapy for non-Hodgkin
lymphoma in children with primary immunodeficiency: Analysis of 19 patients
from the BFM trials; MEDICAL-AND-PEDIATRIC-ONCOLOGY. DEC 1999; 33 (6) : 536-544
Schneider-DT;
Calaminus-G; Gobel-U-Diagnostic value of alpha(1)-fetoprotein and beta-human chorionic
gonadotropin in infancy and childhood; PEDIATRIC-HEMATOLOGY-AND-ONCOLOGY.
JAN-FEB 2001; 18 (1) : 11-26
Wilda-M;
Demuth-I; Concannon-P; Sperling-K; Hameister-H.Expression pattern of the
Nijmegen breakage syndrome gene, Nbs1, during murine development;
HUMAN-MOLECULAR-GENETICS. JUL 22 2000; 9 (12) : 1739-1744
Wu-GS;
Burns-TF; McDonald-ER; Meng-RD; Kao-G; Muschel-R; Yen-T; El-Deiry-WS.Induction
of the TRAIL receptor KILLER/DR5 in p53-dependent apoptosis but not growth
arrest;ONCOGENE-. NOV 11 1999; 18 (47) : 6411-6418
Riferimenti [modifica]
Saltate^ Boder, E.
(1985). "Atassia-telangiectasia: una visione d'insieme.». Kroc
Serie Fondazione19:. 1-63 PMID2.415.689.
^ Salta a:un b Savitsky, K
.; Bar-Shira, A .; Gilad, S .; Rotman, G .;Ziv, Y
.; Vanagaite, L .; Tagle, DA; Smith, S .; Uziel, T .;Sfez,
S .; Ashkenazi, M .; Pecker, I .; Frydman, M
.; Harnik, R .; Patanjali, BS; Simmons, A .; Clines,
GA; Sartiel, A .; Gatti, RA; Chessa, L .; Sanal, O
.; Lavin, MF; Jaspers, NG; Taylor, AM; Arlett,
CF; Miki, T .; Weissman, SM; Lovett, M .; Collins, FS; Shiloh,
Y. (23 Giu, 1995). "Un singolo gene atassia telangiectasia con
un prodotto simile a PI-3 chinasi.».Science 268 (5218):.
1749-1753 doi: 10.1126 /science.7792600. PMID7.792.600.
Saltate^ Thompson, D
.; Duedal, S .; Kirner, J .; McGuffog, L .;Ultimo, J
.; Reiman, A .; Byrd, P .; Taylor, M .; Easton, DF (1
Giugno 2005). "Rischi
di cancro e la mortalità in eterozigoti portatori della mutazione
ATM.". Journal of National Cancer Institute 97 (11):
813-22. Doi: 10.1093 / JNCI / dji141.PMID15.928.302.
Saltate^ Renwick, A
.; Thompson, D .; Seal, S .; Kelly, P .; Chagtai, T
.; Ahmed, M .; Nord, B .; Jayatilake, H .; Barfoot, R
.;Spanova, K .; McGuffog, L .; Evans, DG; Eccles, D
.; Breast Cancer Suscettibilità Collaboration, (UK); Easton,
DF;Stratton, MR; Rahman, N. (agosto 2006). "Mutazioni ATM
che causano l'atassia telangiectasia-sono al seno suscettibilità alleli
cancro".. Nature Genetics 38 (8):.
873-5 doi: 10.1038
/ng1837. PMID16.832.357.
Saltate^ Paller, AS; Massey,
RB; Curtis, MA; Pelachyk, JM;Dombrowski, HC; Leickly,
FE; Swift, M. (dicembre 1991)."Lesioni granulomatose cutanee
nei pazienti con atassia telangiectasia-".. Journal of
Pediatrics 119 (6): 917-22. Doi:10.1016 / s0022-3476
(05) 83043-4. PMID1.960.607.
^ Salta a:un b McGrath-Morrow,
SA; Gower, WA; Rothblum-Oviatt, C .; Brody,
AS; Langston, C .; Fan, LL; Lefton-Greif, MA;Crawford,
TO; Troche, M .; Sandlund, JT; Auwaerter, PG;Easley, B
.; Loughlin, GM; Carroll, JL; Lederman, HM (settembre
2010). "Valutazione
e gestione della malattia polmonare atassia telangiectasia-".. Pneumologia
pediatrica45 (9): 847-59. Doi: 10.1002 / ppul.21277. PMID20.583.220.
Saltate^ Lefton-Greif,
MA; Crawford, TO; Winkelstein, JA; Loughlin,
GM; Koerner, CB; Zahurak, M .; Lederman, HM (febbraio 2000). "Disfagia orofaringea e
aspirazione in pazienti con atassia telangiectasia-".. Journal
of Pediatrics 136 (2): 225-31. Doi: 10.1016 / s0022-3476
(00) 70106-5PMID
Saltate^ Farr,
AK; Shalev, B .; Crawford, TO; Lederman, HM;Winkelstein,
JA; Repka, MX (dicembre 2002). "Manifestazioni oculari di atassia
telangiectasia-.". American journal of Ophthalmology 134 (6):
891-6. Doi: 10.1016 / s0002-9394
(02) 01796-8. PMID12.470.759.
Saltate^ Savitsky, K
.; Bar-Shira, A .; Gilad, S .; Rotman, G .; Ziv, Y
.; Vanagaite, L .; Tagle, DA; Smith, S .; Uziel, T
.; Sfez, S .;Ashkenazi, M .; Pecker, I .; Frydman, M
.; Harnik, R .;Patanjali, BS; Simmons, A .; Clines,
GA; Sartiel, A .; Gatti, RA; Chessa, L .; Sanal, O .; Lavin,
MF; Jaspers, NG; Taylor, AM; Arlett, CF; Miki, T
.; Weissman, SM; Lovett, M .; Collins, FS; Shiloh, Y.
(23 Giu, 1995). "Un singolo gene atassia telangiectasia con un
prodotto simile a PI-3 chinasi.».Science 268 (5218):.
1749-1753 doi: 10.1126 /science.7792600. PMID7.792.600.
^ Salta a:un b Kurz,
UE; Lees-Miller, SP (agosto-settembre 2004)."Attivazione di ATM
e vie di segnalazione ATM-dipendente DNA danno-indotta.». Riparazione
del DNA 3 (8-9):. 889-900doi: 10.1016 / j.dnarep.2004.03.029. PMID15.279.774.
^ Salta a:un b Dar, io .; Biton, S
.; Silo, Y .; Barzilai, A. (19 LUGLIO 2006). "Analisi
della telangiectasia mutato atassia-mediata DNA risposta al danno nei
neuroni del cervelletto di topo.". Il giornale di
neuroscienze: la rivista ufficiale della Society for Neuroscience 26 (29):.
7767-74 doi: 10,1523 /
JNEUROSCI.2055-06.2006 . PMID16.855.104.
Saltate^ Guo, Z .; Kozlov, S
.; Lavin, MF; Persona, MD; Paull, TT (22 ottobre
2010). "Attivazione Sportello dallo stress
ossidativo.". La scienza 330 (6003):
517-21. Doi: 10.1126 /science.1192912. PMID20.966.255.
Saltate^ Bakkenist,
CJ; Kastan, MB (Gen 30, 2003). "Danno al DNA attiva ATM
attraverso autofosforilazione intermolecolari e dimero dissociazione.
Nature 421 (6922): 499-506. Doi:10.1038 / nature01368. PMID12.556.884.
Saltate^ Kanu, N
.; Behrens, A. (Nov 15, 2008). "ATMINistrating ATM segnalazione:
regolazione di ATM da ATMIN.». Ciclo cellulare (Georgetown,
Tex.) 7 (22):. 3483-6 doi: 10,4161 /cc.7.22.7044. PMID19.001.856.
Saltate^ Shiloh, Y. (marzo
2003). "ATM e relative proteine chinasi:.
Integrità del genoma
salvaguardia". Nature recensioni Cancer3 (3):
155-68. Doi: 10.1038 / nrc1011. PMID12.612.651.
^ Salta a:un b Barlow, C
.; Hirotsune, S .; Paylor, R .; Liyanage, M .;Eckhaus, M
.; Collins, F .; Silo, Y .; Crawley, JN; Ried, T
.;Tagle, D .; Wynshaw-Boris, A. (12 luglio 1996). "Topi
Atm-carenza: un paradigma di atassia telangiectasia.». Cella 86(1):.
159-71 doi: 10.1016 / S0092-8674
(00) 80086-0.PMID8.689.683.
Saltate^ Spina, AW; Peters,
AH; Xu, Y .; Keegan, KS; Hoekstra, MF;Baltimora, D
.; de Boer, P .; Ashley, T. (dicembre 1997)."ATM e RPA in
meiotica sinapsi cromosomica e ricombinazione.". Nature
Genetics 17 (4):. 457-61 doi:10.1038 / ng1297-457. PMID9.398.850.
Saltate^ Franco, S
.; Alt, FW; Manis, JP (8 Set, 2006). "Percorsi che sopprimono
programmati rotture del DNA di progredire a cromosomiche pause e
traslocazioni.». Riparazione del DNA5 (9-10):.
1030-1041 doi: 10.1016 / j.dnarep.2006.05.024.PMID16.934.538.
Saltate^ Bagley, J .; Singh,
G .; Iacomini, J. (Apr 15, 2007)."Regolamento delle risposte di
stress ossidativo da atassia telangiectasia-mutato è richiesto per la
proliferazione T cellulare.". Journal of Immunology
(Baltimore, Md:. 1950) 178(8): 4757-63 doi: 10,4049 / jimmunol.178.8.4757. PMID17.404.255.
Saltate^ Bredemeyer,
AL; Sharma, GG; Huang, CY; Helmink, BA;Walker,
LM; Khor, KC; Nuskey, B .; Sullivan, KE; Pandita,
TK; Bassing, CH; Sleckman, BP (27 Luglio 2006). "ATM
stabilizza DNA complessi doppio filamento-break durante V (D) J
ricombinazione.". Nature 442 (7101):.
466-70 doi:10.1038 / nature04866. PMID16.799.570.
Saltate^ Bredemeyer,
AL; Huang, CY; Walker, LM; Bassing, CH;Sleckman, BP (15
Ago 2008). "Aberrante
V (D) J ricombinazione atassia telangiectasia mutato linfociti con
deficit dipende fine DNA nonhomologous unirsi.". Journal of
Immunology (Baltimore, Md:. 1950) 181 (4):.
2620-5 doi:10,4049 / jimmunol .181.4.2620. PMID18.684.952.
Saltate^ Callen, E
.; Jankovic, M .; Difilippantonio, S .; Daniel, JA;Chen,
HT; Celeste, A .; Pellegrini, M .; McBride, K .;Wangsa, D
.; Bredemeyer, AL; Sleckman, BP; Ried, T .;Nussenzweig, M
.; Nussenzweig, A. (13 luglio 2007). "ATM impedisce la
persistenza e la propagazione di rotture cromosomiche
neilinfociti.». Cella 130 (1):.63-75 doi:10.1016 / j.cell.2007.06.016. PMID17.599.403.
Saltate^ Shiloh, Y .; Tabor,
E .; Becker, Y. (luglio 1982). "-Colonia capacità di
formazione di fibroblasti cutanei atassia telangiectasia-è un indicatore
della loro precoce senescenza e l'aumento della domanda per i fattori di
crescita.". La ricerca sulle cellule sperimentale 140 (1):.
191-9 doi: 10.1016 / 0014-4827 (82) 90169 -0. PMID6.213.420.
Saltate^ Inomata, K .; Aoto,
T .; Binh, NT; Okamoto, N .; Tanimura, S .; Wakayama,
T .; Iseki, S .; Hara, E .; Masunaga, T .;Shimizu, H
.; Nishimura, EK (12 giugno 2009). "Stress genotossico
abroga il rinnovo delle cellule staminali melanociti innescando la loro
differenziazione.". Cella 137(6):.
1088-1099 doi: 10.1016 / j.cell.2009.03.037. PMID19.524.511.
Saltate^ Stray-Pedersen, A
.; Borresen-Dale, AL; Paus, E .;Lindman, CR; Burgers, T
.; Abrahamsen, TG (novembre 2007). "Alpha fetoproteina è
in aumento con l'età atassia telangiectasia-".. European
journal of neurologia pediatrica: EJPN: rivista ufficiale della Società
Europea di Neurologia Pediatrica 11 (6):.375-80 doi: 10.1016 / j.ejpn.2007.04.001. PMID17.540.590.
Saltate^ Biton, S .; Dar, I
.; Mittelman, L .; Pereg, Y .; Barzilai, A .;Shiloh, Y.
(23 Giu, 2006). "Nucleare atassia telangiectasia-mutato (ATM)
media la risposta cellulare al DNA doppie rotture di filamenti in cellule
simili ai neuroni umani".. Il Journal of Biological
Chemistry 281 (25):. 17482-91 doi:10,1074 / jbc.M601895200. PMID16.627.474.
Saltate^ Herzog, KH; Chong,
MJ; Kapsetaki, M .; Morgan, JI;McKinnon, PJ (15 maggio
1998). "Requisito per Atm in ionizzanti morte cellulare indotta
da radiazioni nel sistema nervoso centrale in via di sviluppo.». Science 280 (5366):
1089-91. Doi: 10.1126 / science.280.5366.1089. PMID9.582.124.
Saltate^ Lee, Y
.; Chong, MJ; McKinnon, PJ (1 set 2001). "Atassia Telangiectasia
mutato-dipendente apoptosi dopo lo stress genotossico in sviluppo del
sistema nervoso è determinata dallo stato la differenziazione
cellulare.". Il giornale di neuroscienze: la rivista
ufficiale della Society for Neuroscience 21 (17):.
6687-93 PMID11.517.258.
Saltate^ Iourov,
IY; Vorsanova, SG; Liehr, T .; Kolotii, AD; Yurov, YB
(15 Luglio 2009). "Una maggiore instabilità cromosomica
sconvolge in modo drammatico l'integrità del genoma neurale e media
cerebellare degenerazione del cervello l'atassia
telangiectasia-".. Umana Genetica Molecolare 18 (14):
2656-69. Doi: 10.1093 / HMG / ddp207. PMID19.414.482.
Saltate^ Barzilai, A
.; Biton, S .; Shiloh, Y. (1 luglio 2008). "Il ruolo
del DNA risposta danni nello sviluppo neuronale, l'organizzazione e la
manutenzione.». Riparazione del DNA 7(7):
1010-27. Doi: 10.1016 / j.dnarep.2008.03.005.PMID18.458.000.
Saltate^ Biton, S
.; Barzilai, A .; Shiloh, Y. (1 luglio 2008). "Il
fenotipo neurologico di atassia telangiectasia-: risolvere un puzzle
persistente.". Riparazione del DNA 7 (7):
1028-38.Doi: 10.1016 / j.dnarep.2008.03.006. PMID18.456.574.
Saltate^ Yang, Y
.; Herrup, K. (Mar 9, 2005). "La perdita di controllo del ciclo cellulare
neuronale atassia telangiectasia-:. Un meccanismo di malattia
unificata". Il giornale di neuroscienze: la rivista
ufficiale della Society for Neuroscience 25 (10):.
2522-9 doi: 10,1523 /
JNEUROSCI.4946-04.2005 .PMID15.758.161.
Saltare^ "accumulo nucleare
di HDAC4 in carenza di ATM promuove neurodegenerazione in
atassia-teleangectasia.». Nat Med 18(5):
783-90. Maggio 2012. doi: 10.1038 / nm.2709.PMID22.466.704.
Saltate^ "ATM e le
epigenetica del genoma neuronale.». Mech invecchiamento Dev 134 (10):
434-9. Ottobre 2013. doi:10.1016 / j.mad.2013.05.005. PMID23.707.635.
Saltate^ Gatti, RA; Berkel,
I .; Boder, E .; Braedt, G .; Charmley, P
.; Concannon, P .; Ersoy, F .; Foroud, T .; Jaspers,
NG;Lange, K. et al. (1988). "Localizzazione di un gene
atassia telangiectasia-al cromosoma 11q22-23". Nature 336 (6199):.
577-580 doi: 10.1038 / 336577a0. PMID3.200.306.
Saltate^ Sole, X
.; Becker-Catania, SG; Chun, HH; Hwang, MJ; Huo, Y
.; Wang, Z .; Mitui, M .; Sanal, O .; Chessa, L
.; Crandall, B .; Gatti, RA (giugno 2002). "La
diagnosi precoce di atassia telangiectasia-utilizzando test
radiosensitivity.". Journal of Pediatrics 140 (6):
724-31. Doi: 10,1067 /mpd.2002.123879. PMID12.072.877.
Saltate^ Chun, HH; Sole, X
.; Nahas, SA; Teraoka, S .; Lai, CH;Concannon, P
.; Gatti, RA (dicembre 2003). "Migliorata test diagnostici
per l'atassia telangiectasia-by immunoblotting di lisati nucleari per
l'espressione della proteina ATM.". Genetica molecolare e il
metabolismo 80 (4): 437-43. Doi: 10.1016 /j.ymgme.2003.09.008. PMID14.654.357.
Saltate^ Anheim, M
.; Tranchant, C .; Koenig, M. (Feb 16, 2012). "Il autosomiche recessive
atassie cerebellari.". Il New England Journal of
Medicine 366 (7): 636-46. Doi: 10,1056 /NEJMra1006610. PMID22.335.741.
L’atassia familiare episodica o
periodica (EA) è una rara affezione ereditaria a trasmissione autosomica
dominante della quale si descrivono attualmente due tipi (Jen et al., 2007):
Ci sono due varianti
comuni della sindrome atassia episodica, chiamati EA1 e EA2, nonché una serie
di altre condizioni varie che hanno atassia episodica come caratteristica
comune.Molte di queste sindromi sono anche altri sintomi neurologici associati,
come disturbi del movimento, problemi muscolari o mal di testa. Maggiori informazioni su ereditata
atassia si trova qui.
— atassia
Episodica di tipo 1 (EA1): è dovuta a mutazione di un gene dei canali del
potassio voltaggio-dipendente (KCNAI/ Kvl.1) nel cromosoma l2pl3. La malattia
ha un esordio solitamente tardivo (tra la .5° e la 7° decade) e si
manifesta con crisi di atassia-vertigine di breve durata (secondi/minuti)
solitamente favorite dall’esercizio fisico. In fase interaccessuale vi sono
talora mioclonie ma non vertigini;
— atassia episodica di tipo 2 (EA2): è dovuta a mutazione di un
gene dei canali del calcio (CACNA1A subunità alfal) di tipo P/Q voltaggio-dipendente
localizzato nel cromosoma 19p che è altamente espresso nel cervelletto. La
malattia può esordire dall’infanzia sino alla sesta decade e si caratterizza
per crisi di atassia-vertigine di lunga durata (ore-giorni). Gli attacchi
possono associarsi a diplopia, acufeni, nausea, vomito, disartria, emiparesi,
parestesie, cefalea e disorientamento spazio-temporale. Obiettivamente durante
la crisi si riscontra un nistagmo rotatorio o più frequentemente verticale (più
spesso verso il basso). Un nistagmo spontaneo o posizionale può essere quasi
sempre evidenziato anche in periodo intercritico. Dopo qualche anno di crisi
episodiche di atassia si può determinare in questi soggetti una condizione non
distinguibile dalla SCA6. La terapia con acetazolamide è molto efficace sia nel
ridurre od eliminare gli attacchi che nel prevenire la comparsa di atassia
persistente (Brandt e Strupp, 1997).
Le atassie episodiche sono
molto rari. Nella nostra banca dati clinica dei pazienti vertigini,
contenenti circa 12.000 pazienti distinti con vertigini, abbiamo solo 23
pazienti con diagnosi di "atassia episodica".
Recensione:
Nel 1946, Parker ha
descritto una atassia familiare caratterizzata da attacchi di generalizzata
atassia cerebellare a partire dal 3 ° e 4 ° decennio. Vertigo è
infrequente e sia il tronco e le estremità sono atassica. Possono
verificarsi diplopia e macchiatura di parola. Acetazolamide è stato
trovato per evitare che l'atassia episodica da Griggs ed altri in una famiglia,
e questo farmaco è anche stato successivamente trovato per essere utile a
entrambe le varianti (vedi in seguito così come questo link)
In una seconda
descrizione, Farmer e Mustian (1963) descrissero una famiglia con ricorrenti
vertigini / squilibrio a partire dal decennio 2-4th, seguito da un tronco
atassia lentamente progressiva. Questa sindrome vestibolocerebellare è
dominante ereditato. E 'caratterizzata da atassia episodica (ovviamente), rimbalzo
nistagmo, saccadico overshoot dismetria, l'esercizio anomalo e nistagmo
optocinetico, e normali risposte canale semicircolare. Circa il 50% dei
pazienti riferiscono sintomi di emicrania. Un aspetto sottile di risposte
relative otoliti, la componente di polarizzazione di Ovar, è ridotta in questi
pazienti (Furman et al, 1997). Questo disturbo è legato al cromosoma
19p. Il gene anormale codifica apparentemente una subunità del canale del
calcio, e questi pazienti hanno una diminuzione della loro densità di canali di
calcio. Una mutazione in questo gene è anche associata con emiplegica familiare, nonché in SCA-6
(vedere la successiva), ma non è coinvolto nella variante familiare benigna
vertigini ricorrenti. Questa sindrome atassia episodica risponde Acetazolamide
come la prima variante. Un'altra famiglia con una mutazione nel gene del
canale del calcio 19p con atassia progressiva è stata descritta da Yue et al
(1997).
Brandt e Strupp (1997)
discutono una nomenclatura di EA-1 ed EA-2. EA-1 è senza vertigini, mentre
EA-2 ha le vertigini.
EA-1 è dovuta a
mutazioni in un gene KCNA1 canale del potassio su 12p13. EA1 può iniziare
nella prima infanzia con attacchi di atassia della durata di minuti e con
myokymia interictale.
EA-2 (Atassia episodica,
tipo 2) - mutazione in CACNA1
EA-2 è causata da
mutazioni di un gene CACNL1A4 canale del calcio sul cromosoma 19p, molto
espresso nel cervelletto. EA2 può essere particolarmente importante a causa del suo
legame di emicrania associata vertigini (vedi in seguito), che è comune e può
avere sovrapposizioni sintomatologia. Secondo Baloh e altri (1997), EA2 è
caratterizzata da atassia progressiva con interictale nistagmo rebound evocato
dallo sguardo, così come downbeating nistagmo. Almeno 21 le mutazioni
CACNA1A distinti sono stati identificati in EA2. (Subramony et al,
2003). Alcune varianti hanno anormale fissazione oculare e sensibilità al
calore, simile a quella della sclerosi multipla. Altri fattori scatenanti
possono includere l'etanolo, caffeina o movimenti improvvisi. Nella nostra pratica a CDH, abbiamo sospettato
EA2 in molti pazienti, ma raramente abbiamo ottenuto dati genetici schiena che
conferma il nostro sospetto. Questo è forse dovuto alla mancanza di
beneficio per i pazienti percieved nel fare questo test genetici un po
'costoso, quando le opzioni di trattamento sono così limitati. Tuttavia,
abbiamo un numero di pazienti che hanno fatto bene con il trattamento
(acetazolamide o 4AP).
Complessivamente, questi
disturbi assomigliano il fenotipo di emicrania associata vertigini, e forse c'è una
sovrapposizione tra queste due condizioni. Baloh anche riferito 4 casi di
EA2, con l'emicrania classica, legata al cromosoma 19p (Baloh et al,
1997). Emicrania emiplegica familiare è stata anche legata a mutazioni nel
gene del canale del calcio (Ophoff et al, 1996). Strupp ed altri (2004)
hanno recentemente riportato che EA2 può essere trattata con il bloccante del
canale del potassio 4 aminopiridina (4-AP)
Ancora un'altra sindrome
correlata è causata da una mutazione in CACNA1A. E 'caratterizzata da
atassia episodica e la debolezza o emiplegia. (Jen et al, 1999) Alcuni
parentele hanno myotonia in aggiunta (Zasorin et al, 1983). Un'altra
canalopatia calcio è ipokaliemica paralisi periodica che viene codificata da
mutazioni in CACNA1S. I pazienti con canalopatie calcio tra cui SCA-6 e
EA2 hanno risposte oculari carenti di otoliti ingresso. Questi disturbi
possono anche presentare sintomi molto simili posizionali una condizione
orecchio comune - Vertigine parossistica posizionale benigna (VPPB) (Jen et al, 1998).
Baloh anche riferito 4
casi di EA2, con l'emicrania classica, legata alla chormosome 19p (Baloh et al,
1997).
Emicrania emiplegica
familiare, è in qualche modo simile, ma aggiunge un mal di testa e
emiplegia alla foto. Anche in questo caso ereditaria dominante, esordio è
nell'infanzia o nell'adolescenza. L'emiplegia è tipicamente transitorio,
ma non necessariamente. Nistagmo, può verificarsi atassia e atrofia
cerebellare. Impaired P / Q-canale funzione tipo di calcio è l'eziologia e
la mutazione è di nuovo in CACNL1A (Ophoff et al, 1996).
Uno dei numerosi per
contare atassie spinocerebellari, SCA6, è causa di
una ripetizione a tre nucleotidi su questo gene.
EA3
Un'altra variante, che
si trova in gran parte in North Carolina è "periodica atassia
vestibulocerebellar", un altro autosomica dominante atassia con difettoso
esercizio regolare. Questo disturbo è geneticamente distinto da SCA
1,2,3,4,5 così come atassia episodica con myokymia (12p), nistagmo (19p),
acetazolamide reattiva (19p), e DRPLA (12p), secondo Damji et al ( 1996). L'acronimo
"EA3" è stato suggerito per questa sindrome (Steckley et al,
2001). Questa sindrome non risponde a acetazolamide, non ha myokymia
interictale, e hanno anche i movimenti oculari anomali.
EA4
Una quarta variante
distinta da EA1 e EA2 è stato proposto di essere EA4. Questo disturbo
include episodica atassia vestibolare, vertigini, acufeni, e miochimia
interictale.Così questo disturbo ha sintomi molto somiglianti a malattia di
Meniere. E 'anche acetazolamide reattivo (Steckley et al, 2001). E
'stato descritto in un grande gruppo di affini canadese del patrimonio
mennonita. Dodici dei 26 individui avevano miochimia.
Francese canadese
episodica atassia
I episodica atassia di
volte sono chiamati "francese canadese episodica
Atassia". Questo non è il nome migliore - come non si trova in
franco-canadesi solo -ma è utile ai medici in Nord America come ricorda una
delle grandi tribù che sono stati studiati in Quebec con questa
condizione. Perché francesi canadesi sono un piccolo gruppo che matrimoni
misti, hanno una prevalenza molto più elevato di alcune condizioni genetiche
(Musselman et al, 2014). Un medico canadese, il dottor Adrian Barbeau,
condurre uno sforzo immenso per quanto riguarda un altro atassia autosomica
- Friedreichs atassia, e aveva anche
alcuni commenti a proposito delle atassie episodiche. (es Barbeau e Roy,
1984). Le atassie canadesi francesi sono in gran parte autosomica
recessiva, a differenza della situazione con EA2 (dominante). Ciò
significa che nessuno dei genitori può essere un vettore.
Altri sei tribù, che
rappresentano tre sindromi distinte, sono stati descritti da Gancher e Nutt
(1986). Un gruppo di affini incluse parossistica choreoathetosis.
Diverse varianti
di emicrania e vertigini
episodica sono stati riportati da Baloh. Recentemente si è un rapporto di
un vestibulopathy familiare composto da vertigine episodica e
emicrania. Documenti esami vestibolari profonda perdita vestibolare
bilaterale. Questa sindrome risponde a Acetazolamide (Baloh et al,
1994). Riportato anche da Baloh e soci, una forma esiste con vertigini
episodica e tremore essenziale. Questa forma è anche sensibile alle
acetazolamide. (Baloh et al, 1996).
Il trattamento di
atassia episodica e patologie correlate:
Allo stato attuale
(2014), le principali opzioni per il trattamento delle atassie episodiche sono
acetazolamide e 4-AP.
Yue et al hanno
ipotizzato che il meccanismo per effetto di acetazolamide è di diminuire pH che
inibisce ione permeazione attraverso canali del calcio aperte.Acetazolamide può
stabilizzato canali che non riescono a inattivare
correttamente. Acetazolamide potrebbe non funzionare in tutti i casi, se
la mutazione distorto la regione del poro del canale, alterando l'effetto
stabilizzante di ioni (Yue et al, 1997), H +.
Vedere la 4 pag -AP per ulteriori
informazioni su questo farmaco. E 'più difficile prescrivere ed è
generalmente usata quando acetazolamide non è possibile o utile. 4-AP è un
bloccante dei canali del potassio. E 'sconcertante che dovrebbe funzionare
nelle varianti EA che non riguardano il canale del potassio.
Anche se non c'è
letteratura che conosciamo relativa uso off-label di verapamil e clonazepam,
noi spesso cerchiamo questi farmaci in pazienti con EA pure.
COSA PUOI FARE PER
AIUTARE SE AVETE UN EPISODICA ATAXIA SINDROME?
Volontario o comunque sostenere
gli sforzi per fare ricerche o diffondere le informazioni sulla tua
malattia
Considerate permettendo al
cervello di essere l'autopsia in caso di decesso.
Vedere un neurologo annuale per
documentare il decorso clinico. Tenere un registro accurato su di te
e la tua genealogia.
Immagini:
Immagine di downbeating
nistagmo in EA sopra hanno contribuito da Dr.Dario Yacovino
RIFERIMENTI
REFERENCES
Baloh RW, Winder A.
Acetazolamide-responsive vestibulocerebellar syndrome: clinical and
oculographic features. Neurology
1991:41:429-433
Baloh RW, Jacobson
KJ, Fife T. Familial vestibulopathy: a new dominantly inherited
syndrome. Neurology
1994:40:20-25
Baloh RW, Foster CA,
Yue Q, Nelson SF. Familial migraine with vertigo and essential
tremor. Neurology
1996, 46(2), 458-60
Baloh RW, Yue Q,
Furman JM, Nelson SF. Familial episodic ataxias: clinical
heterogeneity in four families linked to chromosome 19p.
Barbeau, A., M. Roy,
et al. (1984). "Recessive ataxia in Acadians and
"Cajuns"." Can
J Neurol Sci 11(4 Suppl): 526-533.
Brandt T, Strupp M.
Episodic ataxia type 1 and 2 (familial periodic ataxia/vertigo). Audiol Neurootol 2(6):363-83,
1997
Furman
JM. Otolith-ocular responses in familial episodic ataxia linked to
chromosome 19p. Ann
Neurol 1997:42:189-193
Jen J and
others. A novel nonsense mutation in
CACNA1A causes episodic ataxia and hemiplegia.Neurology 1999 53:34-37
Musselman, KE, et
al. (2014). "Prevalence of ataxia in children: a systematic
review." Neurology
82(1): 80-89.
Ophoff RA and
others. Familial hemiplegic migraine and episodic ataxia type-2 are
caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996;87;543-552
Parker
HL. Periodic ataxia. Mayo Clin Proc 1946:38:642-645
Steckley JL and
others. An autosomal dominant disorder with episodic ataxia, vertigo
and tinnitus. Neurology
2001:57:1499-1502
Subramony SH and
others. Novel CACNA1A mutation causes febrile episodic ataxia with
interictal cerebellar deficits. Ann
Neurol 2003:54: 725-31
Strupp M and
others. Treatment of episodic ataxia type 2 with the potassium
channel blocker 4-aminopyridine. Neurology 2004: 6: 1623-1625
Yue Q, Jen JC, Nelson SF, Baloh
RW. Progressive ataxia due to a missense mutation in a calcium
channel gene. Am J. Hum. Gen 61(5):1078-87, 1997
La sindrome dell'X fragile (o sindrome
di Martin-Bell o FRAX)E’
una condizione genetica ereditaria dovuta solitamente alla mutazione completa
del gene FMR1(Fragile X Mental Retardation 1) posizionato sul braccio
lungo del cromosoma X che presenta una rottura(il
sito fragile) dove c’è il gene. Questo gene ha un’importante funzione di regolazionesugli
altri geni mediante la produzione della proteina FMRP (fragile
X Mental Retardation Protein) di cui si parlerà in seguito.
La sindrome dell’X fragile è stata
descritta la prima volta nel 1943 dal neurologo inglese J.P.Martine, dalla
genetista inglese J.Bell e di nuovo, molti anni dopo, dal ricercatore americano
H.Lubs. Solo però nel 1991 diversi studiosi contemporaneamente scoprirono che
la sindrome è causata dalla mutazione del gene FMR1 localizzato in
corrispondenza del sito fragile.
La sindrome è ritenuta la seconda causa
più frequente di disabilità intellettiva con origine genetica dopo la sindrome
di Down e colpisce circa 1 maschio su 4000 ed 1 femmina su 6000. Dal 2001 è
inclusa nell’elenco delle malattie rare stilato dal Ministero della Salute. è
una malattia genetica umana causata dalla mutazione del gene FMR1 sulcromosoma X, mutazione presente
in un maschio su 4000 e in una femmina su 6000. Circa 1 su 256 donne sono portatrici di X-Fragile
e possono trasmetterlo ai loro figli. Circa 1 su 800 maschi sono affetti da
Sindrome dell'X-Fragile; le loro figlie saranno, a loro volta, portatrici del
gene. Si contende con la sindrome di Down il primato come
causa genetica più comune di ritardo mentale (si hanno
comunque casi, anche in Italia, di soggetti affetti da X fragile che hanno
frequentato l'università).
Normalmente il gene FMR1 contiene tra 6 e
53 ripetizioni del codone CGG (ripetizioni
di trinucleotidi). Negli individui affetti dalla sindrome dell'X fragile, l'allele FMR1 ha più di
230 ripetizioni di questo codone. Questo grado di espansione provoca la metilazione delle citosine
nel promotore del gene FMR1, con conseguente silenziamento dell'espressione del
gene FMR1. La metilazione del locus FMR1, che è situato nella
banda cromosomica Xq27.3, provoca in quel punto la costrizione e la fragilità
del cromosoma X, fenomeno che
dà il nome alla sindrome.
I maschi portatori di un gene FMR1 con
una significativa espansione del tripletto CGG presentano i sintomi della
malattia, visto che normalmente possiedono una sola copia del cromosoma X. Le
femmine, invece, possiedono due copie del cromosoma X e pertanto hanno una
probabilità doppia di possedere almeno un allele funzionante. Le donne
portatrici di un gene FMR1 espanso su di uno solo dei due cromosomi X possono
presentare alcuni sintomi della malattia o essere normali.
A parte il ritardo mentale di grado
variabile da moderato a severo, altre evidenti caratteristiche della sindrome
sono il volto allungato, grandi orecchie, grossi testicoli (macrorchidismo), e basso tono muscolare. Le caratteristiche comportamentali
possono comprendere movimenti stereotipati (ad esempio, battere le mani) e
sviluppo sociale atipico, in particolare timidezza e limitato contatto con gli
occhi dell'interlocutore. Alcuni individui affetti dalla sindrome dell'X
fragile rientrano inoltre nei criteri diagnostici dell'autismo.
La mutazione e metilazione del gene FMR1
porta all'abolizione della produzione della proteina per cui il gene FMR1
codifica, detta FMRP (fragile X-mental retardation protein). FMRP è una
proteina legante gli RNA (RNA-binding protein) espressa soprattutto nei
testicoli e nel cervello, i tessuti più colpiti dalla sindrome. FMRP si associa
ad RNA messaggeri codificanti importanti proteine neuronali, e ne regola alcuni
aspetti essenziali, quali il trasporto lungo i dendriti verso le sinapsi e la
traduzione in proteine. In assenza di FMRP, molti degli RNA messaggeri
bersaglio della proteina sono deregolati e sono maggiormente tradotti in
proteina. Emergono inoltre nuove funzioni molecolari della proteina, quali la
regolazione della stabilità degli RNA messaggeri.
Anche se non esiste ancora una cura per
la sindrome, c'è la speranza che una migliore comprensione delle sue cause
possa portare a nuove terapie. Al momento, la sindrome può essere trattata
attraverso una terapia del comportamento, un'educazione speciale, e quando
necessario, con un trattamento delle anomalie fisiche. Si consigliano le
persone che hanno dei parenti affetti dalla sindrome dell'X fragile di contattare
dei genetisti per valutare la probabilità di avere figli malati, e per
conoscere la gravità dei problemi che potrebbero presentare i discendenti
affetti dalla sindrome.
Sindromi correlate
Sindrome tremore/atassia FXTAS
Si manifesta generalmente tra i 50 e gli
80 anni. I sintomi che i familiari possono notare, purtroppo spesso attribuiti
all’invecchiamento, sono: tremori “intenzionali”, ovvero tremori che
intervengono durante un movimento volontario come il tentativo di afferrare un
oggetto, versare da bere oppure scrivere; problemi di equilibrio (atassia), che
causano cadute o instabilità mentre si cammina; addormentamento delle estremità
(neuropatia periferica); cambiamenti d’umore, irritabilità ed altre alterazioni
della personalità; perdita della memoria a breve termine e progressivo declino
intellettivo.
Concetti
generali di Genetica
Le
cellule che compongono l’organismo hanno un nucleo nel quale si trovano i cromosomi.
I cromosomi sono costituiti da DNA (acido desossiribonucleico) formato da 2
filamenti avvolti in forma di doppia elica, costituiti da nucleotidi a loro
volta composti da 1 molecola di acido fosforico, 1 di desossiribosio (zucchero)
e da 4 basi azotate (adenina, timina,citosina e guanina). L’informazione
genetica è determinata dalla sequenza di queste basi azotate che se codificanteviene
chiamatagene. Se un gene è alterato si parla di mutazionee
questa può essere trasmessa dai genitori ai figli.
L’uomo
possiede 46 cromosomi a due a due morfologicamente uguali. Il corredo
cromosomico è costituito quindi da 23 coppie, delle quali un elemento è di
origine paterna e uno di origine materna. I due cromosomi di ogni coppia sono
detti omologhi. Soltanto una coppia è costituita da cromosomi diversi per
morfologia (forma) e funzione: è la coppia da cui dipende la determinazione del
sesso. Questi 2 cromosomi sono detti eterocromosomi nella
femmina sono uguali XX e diseguali nel maschio XY. Le altre coppie di cromosomi
omologhi si chiamano autosomi.
Caratteristiche
Genetiche
Nella
sindrome dell’X fragile l’alterazione genetica (mutazione) consiste nell’amplificazione della
sequenza CGG (citosina-guanina-guanina) che si trova nella porzione iniziale
del gene FMR1. Per semplificare si può dire che nel quadro normalequesta
tripletta è ripetuta circa 5-55 volte e viene trasmessa stabilmente attraverso
le generazioni, nel quadro intermedio o dipremutazioneè
ripetuta 55-200 volte e nella mutazione completa (malattia)
più di 200 volte.
In
presenza di una mutazione completa si ha una metilazione delle citosine
presenti nel DNA che blocca la funzione del gene e quindi non si ha produzione
della proteina FMRP. Questa proteina ha una funzione molto importante specie a
livello dellesinapsi, cioè quelle connessioni che ci sono fra le
cellule nervose (neuroni), che si scambiano sostanze chimiche come il
glutammato. Se manca la proteina FMRP le sinapsi diventano iperattive
rispondendo in modo eccessivo al glutammato e funzionando male. Inoltre questa
proteina regola la plasticità delle sinapsi importante per la
memoria e l’apprendimento.
L’argomento
è di estrema complessità per cui non mi addentro, vorrei solo
ricordare che nella popolazione generale le femmine portatrici di premutazione
sono circa 1 su 250 mentre i maschi 1 su 800. Le premutazioni non causano la
sindrome dell’X fragile ma si possono associare a:
·FXTAS (Fragile X Tremor Ataxia
Syndrome) cioè sindrome di tremore atassia associata all’X fragile che si
manifesta specie nei maschi dopo i 50 anni;
·FXPOI insufficienza ovarica precoce
associata all’X fragile che comporta menopausa prima dei 40 anni.
Quindi,
in conclusione, la mutazione o la premutazione vengono trasmesse dai genitori
ai figli come carattere legato al cromosoma X, occorre però ricordare che il
padre portatore di premutazione la trasmetterà solo alle figlie femmine in
quanto al figlio maschio trasmette il cromosoma Y (la premutazione è sul
cromosoma X).
Diverso è per la madre portatrice di premutazione in
quanto per ragioni non perfettamente chiare la premutazione è altamente
instabile e nella trasmissione può espandersi fino alla condizione di mutazione
completa. Le portatrici di premutazione hanno il 50% di probabilità in ogni
gravidanza di trasmettere la premutazione ai figli sia maschi che femmine
Quadro
Clinico
Le manifestazioni
cliniche della malattia sono più evidenti nei maschi, le femmine
possono avere anche un aspetto normale per cui la diagnosi è per loro a
volte tardiva, le più frequenti sono:
Come
già detto la sindrome dell’X fragile è la causa più frequente di disabilità
intellettiva con origine genetica dopo la sindrome di Down. La disabilità
intellettiva può essere di grado variabile si va da casi lievi con
difficoltà di apprendimento a casi molto gravi di ritardo marcato nello
sviluppo intellettivo.
Altro
disturbo importante è la disabilità nell’apprendimento.
La
sindrome dell’X fragile è difficile da diagnosticare. La diagnosi
può essere sospettata in età pediatrica in bambini che presentano ritardo
psicomotorio aspecifico associato a ritardo nello sviluppo del linguaggio,
disturbi comportamentali (specie difficoltà relazionale) o che abbiano
familiarità per disabilità intellettiva. Alla nascita di solito il bambino non
presenta segni che facciano sospettare la presenza della sindrome. Molti autori
sostengono che l’età media alla quale si arriva alla diagnosi si è abbassata
nel corso degli anni ma purtroppo non si sono ancora raggiunti i risultati
ottenuti negli Stati Uniti.
Come
si effettua
Per
effettuare la diagnosi della sindrome dell’X fragile si ricorre ad un’ indagine
molecolare del DNA (con un semplice prelievo di sangue) che viene
effettuata sul gene FMR1 e consente di stabilire sia l’espansione anomala della
tripletta CGG che lo stato di metilazione del gene. In questo modo possono
essere identificate sia le persone affette dalla sindrome che
i portatori.
L’analisi
è complessa e deve essere effettuata in centri specializzati. Se le
donne che si sono sottoposte al test molecolare sono risultate portatrici di
premutazione o mutazione completa viene proposta loro la diagnosi
prenatale per verificare l’eventuale trasmissione della mutazione al
feto. L’esame viene effettuato sul DNA del feto ottenuto con villocentesi (prelievo
dei villi coriali fra la 10 e la 12 settimana di gestazione) o amniocentesi (prelievo
del liquido amniotico fra la 15 e la 19 settimana di gestazione). Non mi
soffermo sulle modalità del test e sui tempi di esecuzione).
Un
altro tipo di indagine è la diagnosi preconcezionale (Preconceptional
Genetic Diagnosis PCGD) cioè una particolare tecnica che consente di
identificare una mutazione di origine materna prima che avvenga il concepimento.
La tecnica è complessa e ha importanti limiti.
Importante
la diagnosi genetica preimpianto (Preimplantation genetic
Diagnosis PGD) che è una procedura che serve ad individuare anomalie genetiche
negli embrioni ottenuti con fecondazione in vitro. Viene eseguita al terzo
giorno dalla fecondazione su embrioni di 7-8 cellule. Gli embrioni vengono
sottoposti a biopsia e quindi il il loro DNA analizzato per ricercare la
mutazione del gene FMR1. Se il DNA non presenta l’alterazione specifica
l’embrione risulta sano, viene trasferito in utero e inizia la gravidanza. Tale
tecnica, come è ben comprensibile, pone dei problemi di ordine etico.
La diagnosi per FXTAS è positiva se nel paziente
vengono riscontrati: 1) i sintomi appena descritti durante un esame neurologico;
2) la premutazione attraverso il test del DNA; 3) segni compatibili con FXTAS
alla risonanza magnetica, come ad esempio lesioni della sostanza bianca del
cervello (tipica è l’iperintensità in T2 dei peduncoli cerebellari medi) oppure
atrofia generalizzata della corteccia.
La FXTAS è una condizione progressiva, che inizialmente
si può manifestare con sintomi piuttosto lievi che si aggravano col passare del
tempo. Ad ogni modo, la progressione della malattia può variare molto da
persona a persona. Molti pazienti mantengono una buona funzionalità per anni,
persino per decenni, fino a quando diventa difficile eseguire dei compiti
giornalieri oppure camminare senza sostegno. Altri pazienti mostrano solo
tremore e/o atassia e non hanno disturbi cognitivi o psichiatrici. I portatori
della premutazione del gene FMR1 oltre i 50 anni di età sono a rischio di
FXTAS. La condizione, al momento, non è mai stata riportata in pazienti con la
mutazione completa. Inoltre, la FXTAS si manifesta più frequentemente e con
maggiore gravità negli uomini. Gli esperti ritengono che almeno un terzo dei
portatori di sesso maschile sia a rischio di sviluppare i sintomi. Ancora non è
disponibile una cura per la FXTAS, ma i sintomi possono essere trattati per
ridurne la gravità e la progressione. Specifici farmaci possono dunque essere
prescritti per alleviare i tremori e i sintomi fisici e psichici. La terapia
occupazionale, la psicoterapia e la fisioterapia possono essere d’aiuto.
La FXTAS è spesso confusa in sede diagnostica con altre
condizioni, tra cui il morbo di Parkinson, il morbo di Alzheimer, la demenza,
l’ictus e la neuropatia periferica. Chiunque manifesti i sintomi sopra indicati
dovrebbe contattare il proprio medico di famiglia e richiedere il consulto con
un neurologo. Alcuni medici e neurologi potrebbero non essere a conoscenza di
questa sindrome perché di recente scoperta (2001), ma possono trovare
informazioni esaurienti sul sito www.xfragile.net.
Insufficienza ovarica precoce FXPOI
È una condizione che si manifesta con una ridotta o
anomala attività ovarica. Tra le sue conseguenze ci sono forme di infertilità o
“subfertilità”, cicli mestruali irregolari o assenti, “fallimento” ovarico
prematuro (POF) con associata menopausa ed anomalie nei livelli ormonali
(innalzamento dell’FSH). Per POF si intende l’interruzione delle mestruazioni
prima dei 40 anni di età. Rappresenta la forma più grave di FXPOI. Anche donne
non portatrici di premutazione possono andare incontro a insufficienza ovarica
precoce (POI) o “fallimento” ovarico prematuro (POF) per altre cause.
Anche se la FXPOI somiglia alla menopausa per alcuni
sintomi, quali caldane e secchezza vaginale, la FXPOI non equivale alla
menopausa. Infatti, si differenzia per due aspetti importanti: 1) una donna
affetta dalla FXPOI può occasionalmente rimanere incinta, perché le sue ovaie
sono ancora in grado di rilasciare gli ovociti. Al contrario, una donna in
menopausa non può rimanere incinta perché la produzione di ovociti è terminata.
2) una donna con FXPOI potrebbe avere nuovamente dei cicli mestruali, ma non
una donna in menopausa.
Studi clinici hanno dimostrato che il 20-25% delle
donne con una premutazione del gene FMR1 sviluppa la FXPOI.
1) Le donne con premutazione spesso entrano in
menopausa in media cinque anni prima delle non-portatrici.
2) A causa delle alterazioni nei livelli ormonali che
accompagna questa condizione, le donne con FXPOI sono a rischio di osteoporosi
ad un’età più precoce.
3) Le donne portatrici della premutazione non sono
necessariamente meno fertili, per cui è necessario assumere delle precauzioni
contraccettive se non si desidera rimanere incinta.
4) Le donne con la premutazione corrono il rischio di
avere figli con Sindrome X Fragile, che può causare varie forme di disabilità
intellettiva, ritardo nello sviluppo del linguaggio e disturbi comportamentali
(per maggiori informazioni, visitate www.xfragile.net).
5) Poiché sono a rischio di FXPOI, le donne con la
premutazione che desiderino utilizzare la diagnosi genetica pre-impianto (PGD)
a seguito della fecondazione assistita per concepire figli senza Sindrome X
Fragile, dovrebbero verificare la propria idoneità a tali procedure
sottoponendosi ad un test della riserva ovarica (noto anche come test FSH).
6) Le donne con la premutazione potrebbero sviluppare
la FXTAS, anche se questa condizione si manifesta più comunemente negli uomini.
Ad oggi, il rischio per le donne risulta piuttosto modesto.
7) Anche i familiari di portatrici potrebbero
presentare la premutazione. In questo caso, possono andare incontro agli stessi
rischi elencati finora.
Bibliografia
1. Amiri et al. Fragile X-Associated Tremor / Ataxia
Syndrome. Archives of Neurology. VOL 65 (no.
1), gennaio 2008
2.^ Jump up to:Unab Hagerman, PJ; Hagerman RJ (2004). . "Sindrome X-associata tremore / atassia Fragile
(FXTAS)" ritardo mentale e disabilità dello sviluppo Research
Recensioni10 (1):. 25-30 doi:10.1002 / mrdd.20005.
4.^ Hagerman, RJ; Hall, DA, Coffey, S., Leehey, M.,
Bourgeois, J., Gould, J., Zhang, L., Seritan, A., Berry-Kravis, E., Olichney,
J. Miller, JW, Fong, AL, Carpentiere, R., Bodine, C., Gane, LW, Rainin, E.,
Hagerman, H., e Hagerman, PJ (2008). ". Il
trattamento della sindrome X fragile associata tremori atassia (FXTAS) e dei
relativi problemi neurologici". Intervento clinica in Aging3(2):
251-262.
L'atassia di
Friedreich (FA), descritta da Nicholaus Friedreich nel 1863, è l’atassia ereditaria più comune. ,
prima FXTAS è stato
scoperto. L’atassia spino-cerebellare di Friedreichs è una malattia
neurodegenerativa autosomica recessiva con una prevalenza stimata di 1:50000-1:
29000. L’incidenza è molto più bassa negli asiatici e nei soggetti di
discendenza africana. La percentuale dei portatori è stata stimata a
1:120-1:60. Prevalenza si stima in popolazioni europee è di 1 su
50.000. La malattia è causata dall’espansione ripetuta di un
trinucleotide, cioè dall’espansione di una tripletta di GÀA localizzata
all’interno del primo introne del gene della fratassina nel cromosoma 9q
,l’espansione provoca un difetto prescrizionale del gene fratxin, una piccola
proteina mitocondriale, che è responsabile per tutte le manifestazioni cliniche
della FA (Durr et al , 1996). C’è una correlazione chiara tra dimensione della
ripetizione espansa e la gravità del fenotipo in FRDÀ. La fratassina è una
proteina mitocondriale che ha un ruolo nell’omeostasi del ferro La deficienza
di fratassina dà luogo ad accumulo mitocondriale di ferro, a difetti a carico
di enzimi mitocondriali specifici, ad aumentata sensibilità agli stress
ossidativi ed infine alla morte cellulare mediata da radicali liberi Le lesioni
sono rappresentate da fenomeni di tipo degenerativo a carico dei cordoni
posteriori e laterali del midollo spinale, delle strutture del tronco
encefalico compresi i nuclei vestibolari e della corteccia cerebellare.
Considerando
tutti i casi di ereditata atassia, alleli FA sono state trovate in circa il
11,4% dei apparentemente recessiva e il 5,2% dei pazienti apparentemente
sporadici (Moseley et al, 1998).
Una quantità
immensa di lavoro su Friedreichs è stato guidato da Dr. Adrian Barbeau, in
Quebec, Canada.
Epidemiologia
In Europa tale atassia
colpisce mediamente una persona su 50.000 e incide su persone fino alla seconda
decade di età.
Nel 1984 uno studio
canadese ha trovato traccia di 40 casi riconducibili a tale malattia, tutte
persone discendenti dallo stesso ceppo genealogico[Barbeau et al.,1984]. La
validità dei loro studi, nonché altri più grandi è in qualche modo sospetto in
quanto questo lavoro è stato fatto prima della caratterizzazione genetica della
malattia.
Classificazione
Esistono
varie forme di atassia. Alcune di queste sono legate ad alterazioni metaboliche
su base genetica o no, traumatiche ecc. L'atassia di Friedreich è un'atassia
metabolica (nella sua patogenesi sembra implicata la deficienza di una
proteina, la fratassina, che avrebbe il compito di smaltire i metaboliti di
rifiuto dei processi energetici all'interno del mitocondrio), su base genetica:
l'individuo affetto ha 2 geni anomali con un numero esuberante di triplette che
impediscono l'adeguato "srotolamento" del DNA che favorisce la
sintesi della fratassina e che infatti è deficitaria.
Genetica
L'atassia di Friedreich
ha un modello di trasmissione autosomica recessiva.
L'atassia di Friedreich
è una malattia autosomica recessiva che si verifica quando il gene FXN contiene
amplificate intronic ripetizioni GAA (un esempio di espansione ripetizione
Trinucleotide). Il gene FXN codifica per la proteina fratassina. [Klockgether
T 2011)] espansione GAA ripetizione provoca livelli di fratassina di ridurre.
La fratassina è una proteina ferro-legante responsabile per la formazione di
centri ferro-zolfo. Un risultato di carenza di fratassina è sovraccarico di
ferro mitocondriale che può causare danni a molte proteine. [Klockgether T
2011] Il ruolo esatto di fratassina nella fisiologia normale rimane poco
chiaro. [Marmolino, D., 2011] Il gene è localizzato sul cromosoma 9.
Il gene mutante contiene
espanso GAA tripletta ripetizioni nel primo introne; [Montermini et al.,1997]
in alcuni pedigree, mutazioni puntiformi sono stati rilevati. Poiché il difetto
si trova in un introne (che viene rimosso dalla trascrizione mRNA tra trascrizione
e traduzione), questa mutazione non comporta la produzione di proteine fratassina anormali.
Invece, la mutazione causa silenziamento genico (cioè, la mutazione diminuisce
la trascrizione del gene) attraverso l'induzione di un eterocromatina struttura
in un modo simile a effetto di posizione variegato. [ Friedreich Ataxia at eMedicine]
Tale
malattia è causata dalla mutazione di un gene, FRDA o X25 localizzato nel locus
q13-q21 del cromosoma 9 e che codifica per una proteina denominata fratassina.
All’atassia di Friedreich è associata la presenza di un'area di espansione di
triplette nucleotidiche (GAA) ripetute in un introne adiacente al
gene[Campuzano et al.,1996; Montermini et al.,1995].Quello che segue è solo una
breve discussione su come autosomiche recessive ereditarietà funziona. Non è
destinata a sostituire la consulenza genetica.
FA è quasi
sempre una malattia autosomica recessiva. In generale poi, tutti figli di
persone con FA sono portatori FA. Non possono sviluppare FA stessi finché il
loro loro altro parrent non è anche un vettore. Poiché la frequenza di FA è di
1 / 50.000 nella popolazione generale, la possibilità di incontrare un altro
vettore in un coniuge è di circa 1/200.Quando un omozigote per un disturbo
recessivo sposa un vettore, la probabilità di essere colpiti i bambini è del
50%. Così, la possibilità che una persona con i figli di FA avrà anche FA è di
circa 1/400.
Qualora due
vettori si sposano, c'è una probabilità del 25% che il bambino sarà
interessato, una probabilità del 50% che il bambino sarà un vettore, e una
probabilità del 25% che il bambino non porterà la mutazione a tutti. (Bidichandani
e Delatycki 1993)Fratelli di una persona con FA hanno una probabilità del 25%
se stessi di avere FA, una probabilità del 50% di essere un vettore, e una
probabilità del 25% di non avere mutazione.
Oltre a
ridurre l'espressione della fratassina, lunghi tratti di GAA ripete indurre
rotture cromosomiche in in vivo studi di lievito.
Patogenesi
Bassi livelli di
fratassina portano a scarsa biosintesi dei cluster ferro-zolfo che sono
richiesti per il trasporto degli elettroni mitocondriale e montaggio di funzionale aconitasi e dismetabolismo
ferro di tutta la cellula. In individui normali, il gene FXN codifica
fratassina, una proteina della matrice mitocondriale. Questa proteina
globulare consiste di due eliche alfa e sette trefoli beta ed è altamente
conservata, che si verificano in tutti gli eucarioti e procarioti alcuni [Pandolfo, M ( 2008] La fratassina ha
una varietà di funzioni note. Frataxin assiste ferro-zolfo
proteina sintesi
della catena di trasporto degli elettroni per generare definitiva adenosina
trifosfato (ATP), la moneta energia necessaria per svolgere funzioni
metaboliche in cellule. Frataxin regola anche il trasferimento a caldo nei
mitocondri per fornire una adeguata quantità di specie reattive dell'ossigeno
(ROS) per mantenere i processi normali
[Sahdeoé et
al., 2014][. Senza fratassina l'energia nei mitocondri fallisce, e
ferro eccesso provoca ulteriore ROS da creare, portando a ulteriori danni alle
cellule [Pandolfo, M 2008Sahdeoé et al., 2014].
Quadro
clinico:
L’esordio di
solito è erso la pubertà, ma può variare da 2-3 anni di età a oltre 2.5 anni di
età e solitamente è rappresentato da un disturbo della deambulazione di tipo
atassico, che progressivamente si aggrava sino alla perdita della deambulazione
che avviene mediamente dai 7 ai 15 anni dall’esordio della malattia e si va ad
accompagnare ad altri sintomi e segni neurologici che in fase conclamata sono:
riflessi tendinei assenti alle estremità inferiori, evidenza elettrofisiologica
di neuropatia assonale sensitiva seguita da (entro 5 anni dall’esordio)
disartria, areflessia in tutti i quattro arti, perdita distale della
sensibilità di posizione e pallestesica, amiotrofia dei piccoli muscoli della
mano e dei muscoli distali della gamba e del piede, risposta piantare in
estensione(Durr et al, 1996). Più della metà dei pazienti manifesta scoliosi
progressiva, e circa la metà manifesta piede cavo, piede equinovaro e dita dei
piedi ad artiglio. Altri reperti neurologici variabili includono anormalità
oculomotorie, nistagmo, atrofia ottica (25%) e perdita d’udito neurosensoriale
(20%).
In circa i due
terzi dei pazienti vi sono alterazioni cardiache (cardiomiopatia ipertrofica
concentrica) che costituiscono la causa più comune di morte che per tale
patologia può verificarsi anche in età giovanile. Deformità scheletriche e
anomalie nel metabolismo del glucosio sono comuni. Altro risultato
insolito associato può essere il dismorfismo facciale, mioclono, distonia,
tremore posturale, sopranucleare sguardo paresi, e ritardo mentale (Durr et al,
1996)
L’anomalia
del movimento oculare più comune è l’instabilità nello sguardo fisso. Non si
osserva oftalmoparesi. Le alterazioni di competenza otoneurologica sono
costituite in circa la metà dei casi dalla presenza di un nistagmo spontaneo
orizzontale (più raramente verticale verso il basso) o di tipo “alternante”.
Nella maggior parte dei casi la refiettività vestibolare risulta ridotta. Oltre
alla diminuzione dei valori dei parametri quantitativi, il nistagmo provocato
dallo stimolo calorico presenta varie alterazioni qualitative (Eh et al.,1984).
Il sintomo vertigine è riferito raramente ed in genere è di modesta entità,
mentre sono rilevanti i disturbi della postura che si aggravano nettamente con
la chiusura degli occhi. Furman e altri hanno riferito un complesso di sintomi
simile di instabilità fissazione, saccadi imprecisi, esercizio regolare
alterato e una diminuzione del guadagno VOR. Di nuovo, è difficile sapere
se questi pazienti avevano veramente FA, in quanto questo è uno studio più
vecchio. Nella popolazione ben caratterizzato di Durr (1996), nistagmo orizzontale
è stato trovato nel 40%, e di esercizio saccadico nel 30%. A nostro
avviso, questi risultati sono sia quelli soggettivi e comuni in persone
altrimenti normali, forse l'assunzione di farmaci per dormire o bere
alcolici. In altre parole, siamo dubbia che ci sono sostanziali anomalie
dei movimenti oculari in Friedreichs.
La perdita
dell'udito è stata riportata solo nel 13% dei pazienti di Durr
(1996). Spoendlin commentato che due avevano audiogrammi "a
punta". Tuttavia, uditive anormale evocati risposte sono trovati in
circa il 50% dei pazienti, e la sindrome di neuropatia uditiva è stata riportata
in questa malattia. Sembra ragionevole che ci potrebbe essere più di
anormale ABR in questo disturbo di perdita dell'udito, anche se non sembra
essere la causa di danni rilevanti. Secondo Satya-Murti et al (1980),
sentendo disturbo è associato con spirale degenerazione ganglio. Questo è
dunque un altro ganglio sensoriale che degenera. Noi non vediamo il motivo
per cui ci dovrebbe essere alcuna selettività per la perdita di udito, e non
capiamo le osservazioni di Spoendlin (ad esempio, forse sono sbagliate).
Ci si
aspetterebbe che il ganglio vestibolare potrebbe anche degenerare in
Friedreichs, e gli studi patologici hanno suggerito che questo è il
caso. Una situazione simile si trova nella sindrome tela, che è molto
simile a Friedreichs eccetto che ha disturbi più vestibolare. (Szmulewicz
et al, 2011)
La FRDA è
una malattia a carattere progressivo con esito infausto per la quale non
esistono terapie farmacologiche efficaci ma solo provvedimenti di tipo
sintomatico, Questo motivo, unitamente al fatto che esistono moltissimi quadri
cimici che, per lo meno in fase di esordio, possono presentrarsi con una
sintomatologia atassica sostanzialmente sovrapponibile a quella della malattia
di Friedreich, rende indispensabile una diagnostica differenziale
particolarmente scrupolosa al fine di escludere di trovarsi di fronte ad una
delle cosiddette “atassie trattabili” e tra queste soprattutto l’atassia da
deficit isolato di vitamina E (AVED). Si tratta di un’atassia ereditaria di
origine genetica (cromosoma 8Q13, gene codificante il trasportatore dell’alfa
tocoferolo) ad esordio giovanile i cui sintomi sono molto simili a quelli
dell’atassia di Friedreich ma, per la quale, a differenza di questa, per lei
esiste in realtà un trattamento curativo in assenza del quale le lesioni
neurologiche (e la sintomatologia) continuano e si aggravano irrimediabilmente
in modo progressivo.
L’esame dei
livelli plasmatrici di vitamina E è di conseguenza decisivo Ct la diagnosi, per
l’instaurazione della terapia sostitutiva e quindi per la prognosi. Oltre alla
rara AVED esistono diverse altre affezioni “trattabili” che debbono essere
prese in considerazione di fronte ad una sindrome atassica e, limitandoci per
motivi di spazio solo alla loro citazione, ricordiamo principalmente
l’abetalipoproteinemia, la celiachia, la malattia di Refsum, il morbo di
Wilson, la malattia di Wernicke-Korsakoff, la tabe, la pseudotabe alcolica, la
mielosi funicolare da deficit di vitamina B12, la carenza di acido folico,la
xantomatosi cerebro-tendinea, la malattia di Tay-Sachs ad esordio tardivo, la
malattia di Behet, la malattia di Fabry, l’ipotiroidismo, le sindromi
paraneoplastiche, le intossicazioni da ossido di carbonio e da farmaci (litio,
difenilidantoina ecc.).
Prognosi
Nei pazienti
omozigoti per l'espansione GAA, il tempo medio di sedie a rotelle di
confinamento medie 10.8 anni, e la durata media della malattia è di 15 + -9
anni. (COSEE et al, 1999). La morte avviene spesso a causa della
cardiomiopatia.
Patologia:
La patologia
della FA è sostanzialmente limitato al ganglio della radice
dorsale. Neuroni cerebellari sono di solito normali, ma ci sono anche
lesioni nel nucleo dentato. Ci sono anche gli effetti sul cuore, scheletro
e pancreas endocrino. Ossa temporali da pazienti con Freidreichs hanno
dimostrato ganglio spirale e la degenerazione delle cellule gangliari della
Scarpa (Merchant et al, 2001). Circa 2/3 dei pazienti hanno una cardiomiopatia,
e la maggior parte dei pazienti alla fine soccombere ad esso (Bidichandani et
al, 1993). Molti diventano diabetici durante la loro vita (Koeppen AH,
2011).
Trattamento
Una persona che soffre
di Atassia di Friedreich può richiedere alcuni interventi chirurgici
(soprattutto per la colonna vertebrale e il cuore). Spesso, viti e barre
in titanio vengono inseriti nella colonna vertebrale per aiutare a prevenire o
rallentare la progressione della scoliosi. Come si verifica la
progressione di atassia, dispositivi di assistenza, come un bastone,
deambulatore o sedia a rotelle sono necessari per la mobilità e
l'indipendenza. Altre tecnologie assistive, come
ad esempio un telaio in piedi, può
aiutare a ridurre le complicazioni secondarie di uso prolungato di una sedia
a rotelle. Lo
scopo della chirurgia è quello di mantenere il paziente ambulatoriale a lungo
possibile.
In molti casi, i
pazienti sperimentano condizioni cardiache significative pure. Queste
condizioni sono molto più curabile, e sono spesso contrastate con ACE-inibitori, come enalapril o
lisinopril e di altri farmaci cardiaci come digossina.
Le persone con atassia
di Friedreich possono anche beneficiare di un approccio di trattamento
conservativo per la gestione dei sintomi. Gli operatori sanitari istruiti
in condizioni neurologiche, come fisioterapisti e terapisti occupazionali, possono
prescrivere un programma di esercizi su misura per massimizzare la funzione e
l'indipendenza. Per affrontare il modello di andatura atassica e la
perdita di propriocezione tipicamente
visto nelle persone con atassia di Friedreich, fisioterapisti possono
utilizzare cue visivo durante l'andatura formazione per
contribuire a facilitare un più efficiente andatura modello. [15] La prescrizione
di un ausilio con andatura formazione possibile anche prolungare la deambulazione
autonoma. [15]
Esercizi di bassa
intensità rafforzamento dovrebbero essere incorporati per mantenere uso
funzionale degli arti superiori e inferiori. [16] Faticabilità
devono essere attentamente monitorati.Esercizi di stabilizzazione del tronco e
bassa posteriore possono aiutare con controllo posturale e la gestione di scoliosi. [15] Ciò è particolarmente
indicativa qualora la persona non è ambulatoriale e richiede l'uso di una sedia
a rotelle. Equilibrio e formazione coordinamento con feedback visivo
possono anche essere incorporati nelle attività della vita quotidiana. [15]Esercizi dovrebbero
riflettere le attività funzionali quali cucina, trasferimenti e cura di
sé. Insieme con la rieducazione alla deambulazione, l'equilibrio e la
formazione di coordinamento dovrebbe essere sviluppata per ridurre al minimo il
rischio di cadute. [15]
Esercizi di stretching
possono essere prescritti per alleviare la muscolatura stretto a causa di scoliosi e piede
cavo deformità. [16]
Idebenone
Idebenone, un
antiossidante, è stato recentemente rimosso dal mercato canadese nel 2013 a
causa della mancanza di efficacia. [17]
Il trattamento
combinato con Idebenone e deferiprone è stato segnalato utile in 20 pazienti
con FA (Velasco-Sanchez et al, 2011).
C'è una revisione
Cochrane su Antiossidanti e altro trattamento farmacologico in Friedreich che è
stato pubblicato nel 2012 e dovrebbe essere aggiornato entro la fine del
2015. [18]
La somministrazione
Nicotinamide su pazienti è stata associata ad un costante miglioramento
della fratassina concentrazioni verso quelli osservati nei portatori
asintomatici durante 8 settimane di somministrazione giornaliera. La
somministrazione orale giornaliera di 3,8 g nicotinamide ha determinato un
aumento di 1,5 volte, mentre 7,5 g portato a un raddoppio della concentrazione
di proteina fratassina. [21]
Logopedia
I pazienti spesso si
impegnano logopedia da disartria (un disturbo del linguaggio motore) si
verifica in quasi il 100% dei pazienti con atassia di
Friedreich. Logopedia si propone di migliorare i risultati del linguaggio
e / o compensare deficit di comunicazione. [22]
Pensiero
attuale è che non esiste alcun agente che ha un effetto significativamente positivo.
^ Barbeau
A, Sadibelouiz M, Roy M, Lemieux B, Bouchard JP, Geoffroy G, Origin of
Friedreich's disease in Quebec in The Canadian journal of
neurological sciences. Le journal canadien des sciences neurologiques,
vol. 11, 4 Suppl, 1984, pp. 506-9, PMID6391645.
^ Campuzano
V, Montermini
L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius
F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI,
Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F,
Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M, Friedreich's
ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat
expansion in Science, vol. 271, nº 5254, marzo
1996, pp. 1423–7, DOI:10.1126/science.271.5254.1423, PMID8596916
Bidichandani,
SI and MB Delatycki (1993). "Friedreich Ataxia."
Cosee M and
many others. Friedreichs ataxia: point mutations and clinical
presentations of compound heterozygotes. Ann Neurol 1999:45:200-206
De Pablos C,
Berciano J, Calleja J. Brain stem auditory evoked potentials and blink reflex
in Friedreich's ataxia. J. Neurol 1991:238:212-6
Durr and
others. Clinical and Genetic Abnormalities in Patients with Friedreich's
ataxia. NEJM 1996:335:1169-75
Ell J, Prasher
D, Rudge P. Neuro-otological abnormalities in Friedreich's ataxia. JNNP
1984:47:26-32
"FARA Human Trials" (PDF). Friedreich's
Ataxia Research Alliance. 2012. Retrieved2012-03-15.
Jump up^ Friedreich
N (1863). "Ueber degenerative Atrophie der spinalen Hinterstränge"
[About degenerative atrophy of the spinal posterior column]. Arch
Pathol Anat Phys Klin Med (in German) 26 (3–4):
391–419. doi:10.1007/BF01930976.
Jump up^ Friedreich
N (1863). "Ueber degenerative Atrophie der spinalen Hinterstränge"
[About degenerative atrophy of the spinal posterior column]. Arch
Pathol Anat Phys Klin Med (in German) 26 (5–6):
433–459. doi:10.1007/BF01878006.
Jump up^ Friedreich
N (1863). "Ueber degenerative Atrophie der spinalen Hinterstränge"
[About degenerative atrophy of the spinal posterior column]. Arch
Pathol Anat Phys Klin Med (in German) 27 (1–2):
1–26. doi:10.1007/BF01938516.
Jump up^ Friedreich
N (1876). "Ueber Ataxie mit besonderer Berücksichtigung der hereditären
Formen" [About ataxia with special reference to hereditary forms]. Arch
Pathol Anat Phys Klin Med (in German) 68 (2): 145–245. doi:10.1007/BF01879049.
Kearney,
Maria; Orrell, Richard W; Fahey, Michael; Pandolfo,
Massimo; Kearney, Mary (2012)."Antiossidanti e altri trattamenti farmacologici
per Friedreich". La Cochrane Database of Systematic Reviews4: CD007791. Doi: 10.1002 / 14651858.CD007791.pub3. PMID22.513.953.Klockgether T
(August 2011). "Update on degenerative ataxias". Curr. Opin.
Neurol. 24 (4): 339–45. doi:10.1097/WCO.0b013e32834875ba. PMID21734495.
Moseley, ML, KA Benzow,
et al. (1998). "Incidence of dominant spinocerebellar
and Friedreich triplet repeats among 361 ataxia families." Neurology
51(6): 1666-1671.
Pandolfo, M
(ottobre 2008). "Atassia di Friedreich.". Archives of
Neurology 65 (10):. 1296-303 doi: 10.1001 / archneur.65.10.1296. PMID18.852.343.
Pandolfo M
(2009). "Friedreich ataxia: The clinical picture". Journal of
Neurology, 256: (1 Suppl), 3-8, doi:
10.1007/s00415-009-1002-3
"Processi FARA umani" (PDF). Atassia
di Friedreich Research Alliance. 2012. Estratto2012-03-15.
Sahdeo, S .; Scott,
BD; McMackin, MZ; Jasoliya, M .; Marrone, B .; Wulff, H
.; Perlman, SL; Pook, MA; Cortopassi, GA (11 agosto
2014). . "Dyclonine salva carenza di fratassina in modelli animali e
cellule buccali dei pazienti con atassia di Friedreich" Human
Molecular Genetics
Satya-Murti
et al. Auditory dysfunction in Friereich ataxia: result of spiral ganglion
degeneration. Neurology 30:1047-1053, 1980
Szmulewicz,
DJ, JA Waterston, et al. (2011). "Cerebellar ataxia, neuropathy,
vestibular areflexia syndrome (CANVAS): a review of the clinical features and
video-oculographic diagnosis." Ann NY Acad Sci 1233: 139-147.
Velasco-Sanchez, D., A.
Aracil, et al. (2011). "Combined therapy with idebenone and deferiprone
in patients with Friedreich's ataxia." Cerebellum 10(1): 1-8.
Vogel, Adam
P; Folker, Joanne; Poole, Matthew L; Vogel, Adam P (2014). "Il trattamento per disturbi
del linguaggio in Friedreich e altre sindromi atassia ereditaria". La Cochrane Database of
Systematic Reviews 10: CD008953.
Von Hippel-Lindau
disease
(VHL) manifests by angiomas of the retina and hemangioblastomas of the
cerebelum.Malattia
di von Hippel-Lindau (VHL) si manifesta con angiomi della retina e
emangioblastoma del cerebelum. Hemangioblastomas can
also occur elsewhere (see image below). Emangioblastomi può verificarsi
anche altrove (vedi immagine sotto).
Epidemiologia
Colpisce
una persona su 36.000. L'incidenza è maggiore intorno ai 30-40 anni, anche se
può manifestarsi a qualunque età.
Cenni
storici
Il
nome della sindrome è dovuto a coloro che per primi la descrissero:
l’oftalmologo tedesco Eugen Von Hippel e il patologo svedese Arvid Lindau.
Eziologia
La causa di questa malattia è una
mutazione di un gene localizzato sul braccio corto del terzo cromosoma, mappato
in 3p25-26, il quale codifica per la proteina VHL. Quest'ultima, normalmente
collabora con le elonghine B e C[1] per legarsi ai fattori di trascrizione HIF
(Hypoxia Induced Factor) idrossilati dalla presenza di ossigeno, ne provoca
l'ubiquitinizzazione e la distruzione tramite il proteasoma. Il complesso coi
geni HIF è responsabile delle risposte cellulari all'ipossia, fra cui lo
sviluppo di neoangiogenesi e l'induzione della proliferazione cellulare; quindi
VHL si comporta come un oncosoppressore e da questo dato sono facilmente
deducibili le conseguenze cliniche della sua mancanza. Il prodotto genico può
essere totalmente assente (delezioni o mutazioni frameshift) con espressione
anomala di codoni di STOP) oppure inattivo, portando a quadri clinici
differenti.
Inoltre blocca il VEGF
The VHL gene was mapped
to the short arm of Cr3 in 1988 by Seizinger et al (Nature 1988:332:268-269),
and isolated in 1993 by Latif (Science 1993:260:1317-1320). Il gene VHL è stato mappato sul
braccio corto del Cr3 nel 1988 da Seizinger et al (Natura 1988: 332: 268-269),
e isolato nel 1993 da Latif (Science 1993: 260: 1317-1320). Life time risk for renal clear cell carcinoma is 70% and
this is the most frequent cause of death. Rischio tempo di vita per
carcinoma renale a cellule chiare è del 70% e questa è la causa più frequente
di morte. Pheochromocytomas affect 7-20% of cases
and can be bilateral or malignant. I feocromocitomi colpiscono 7-20% dei
casi e possono essere bilaterali o maligni. Non-secretory
pancreatic cell tumors also occur. Tumori non secretoria delle cellule
pancreatiche si verificano anche.
The genetic defect is
in the VHL gene which is a tumor supressor gene. Il difetto genetico è nel gene VHL che è un gene
soppressore del tumore. VHL is inherited in an
autosomal dominant fashion, but two copies of the mutation are needed to cause
the disorder. VHL è ereditato in modo autosomico dominante, ma due copie
della mutazione sono necessari per causare il disturbo. A second mutation can occur during a person's lifetime in a particular
cell, which allows tumors and cysts to develop. Una seconda mutazione
può verificarsi durante la vita di una persona in una particolare cella, che
permette tumori e cisti sviluppare.
Epidemiologia
Malattia VHL ha un'incidenza
di uno su 36.000 nascite. Vi è oltre il 90% penetranza all'età di 65
anni [17] L'età alla
diagnosi varia dall'infanzia all'età 60-70 anni, con una età media dei pazienti
al momento della diagnosi clinica di 26 anni.
Segni e sintomi associati alla
malattia VHL includono mal di testa, problemi di equilibrio e camminare,
vertigini, debolezza degli arti, problemi di visione, e la pressione
alta. Condizioni associate con la malattia VHL comprendono angiomatosi,emangioblastomi,feocromocitoma, carcinoma
a cellule renali,pancreatiche cisti (pancreatica
cistoadenoma sierosa), tumore
sacco endolinfatico, e
cystadenomas bilaterali papillari dei epididimo (uomini) o legamento
largo dell'utero (donne). [5 ] [6] Angiomatosi verifica nel 37.2%
dei pazienti con malattia VHL e di solito si verifica nella retina. Come
risultato, la perdita della vista è molto comune. Tuttavia, altri organi
possono essere colpiti: ictus, attacchi cardiaci, e le malattie cardiovascolari
sono ulteriori sintomi più comuni. [3] Circa il 40% delle malattie
VHL presenta con hemangioblastomas del sistema nervoso centrale e sono presenti
in circa il 60-80%. Emangioblastomi spinale si trovano nel 13-59% delle
malattie VHL e sono specifici perché l'80% si trovano nella malattia di
VHL. [7] [8] Anche se tutti questi tumori
sono comuni nella malattia VHL, circa la metà dei casi si presentano con un
solo tumore tipo. [8]
Ogni cellula del corpo
ha 2 copie di ogni gene. Nella malattia VHL, una copia del gene VHL ha una
mutazione e produce una proteina VHL difettosa (pVHL). Tuttavia, la
seconda copia produce ancora una proteina funzionale. Tumori formano da
solo le cellule in cui è stato mutato la seconda copia del gene. Questo è
noto come l'ipotesi due hit. La mancanza
di questa proteina permette tumori caratteristici della sindrome di von
Hippel-Lindau per sviluppare [11] [12]
Circa il 20% dei casi
di malattia di VHL si trovano in individui senza una storia familiare, noto
come de novo mutazioni. Una mutazione ereditaria del gene VHL è
responsabile per il restante 80 percento dei casi. [7]
30-40% delle mutazioni
nel gene VHL sono costituiti da 50-250kb mutazioni per delezione che rimuovono o
parte del gene o l'intero gene e regioni fiancheggianti del DNA. Il
restante 60-70% delle malattie VHL è causato dal troncamento di pVHL da mutazioni nonsenso, mutazioni INDEL o sito di splice mutazioni. [7]
Proteina VHL
La regolamentazione
dei HIF1α da pVHL. Sotto normali livelli di ossigeno, HIF1α lega
pVHL attraverso 2 residui di prolina ossidrilati ed è polyubiquitinated da
pVHL. Questo porta a sua degradazione attraverso il proteasoma. Durante
l'ipossia, i residui di prolina non sono idrossilata e pVHL non possono
vincolare. HIF1α provoca la trascrizione di geni che contengono
l'elemento di risposta ipossia. Nella malattia VHL, mutazioni genetiche
causano alterazioni alla proteina pVHL, di solito per il sito di legame
HIF1α.
La proteina VHL (pVHL)
è coinvolto nella regolazione di una proteina nota come ipossia fattore
inducibile 1α (HIF1α). Si tratta di una subunità di unheterodimeric fattore di trascrizione che a
normali di ossigeno cellulare livelli è
altamente regolamentato. In condizioni fisiologiche normali, pVHL
riconosce e si lega al HIF1α solo quando l'ossigeno è presente a causa
del posto traslazionale idrossilazione di 2 prolina residui
all'interno della proteina HIF1α. pVHL è una ligasi
E3 che ubiquitinates HIF1α e
provoca la degradazione da parte del proteasoma. In condizioni di
scarsità di ossigeno o in caso di malattia in cui il gene VHL VHL è mutato,
pVHL non si lega a HIF1α. Questo permette la subunità di dimerise con
HIF1β ed attivare la trascrizione di una serie di geni, comprendenti vascolare fattore di crescita endoteliale, piastrinica-fattore
di crescita derivato B,eritropoietina e geni coinvolti
nell'assorbimento del glucosio e metabolismo. [12] [13] Un nuovo romanzo
missenso mutazione nei geni VHL c.194 C> T, c.239 G> A, c.278 G> A,
c.319 C> G, c.337 C> G che porta alle seguenti variazioni p.Ala 65 Val,
p.Gly 80 Asp, p.Gly 93 Glu, p.Gln 107 Glu, p.Gln 113 Glu nella proteina
contribuito a renale carcinoma a cellule chiare. [14]
Diagnosi
La rilevazione di
tumori specifici per la malattia VHL è importante nella diagnosi della
malattia. Negli individui con una storia familiare di malattia di VHL, uno
emangioblastoma, feocromocitoma o carcinoma
renale può essere sufficiente per fare una diagnosi. Come tutte le tumori
associati al morbo di VHL si possono trovare sporadicamente, almeno due tumori
devono essere identificati per diagnosticare la malattia VHL in una persona
senza una storia familiare. [7] [8]
Diagnosi genetica è
utile anche nella diagnosi di malattia VHL. In VHL ereditaria, tecniche di
malattie quali meridionale
blotting e sequenziamento del gene possono essere
utilizzati per analizzare DNA e identificare mutazioni. Questi test
possono essere usati per lo screening familiari di coloro che sono colpiti con
la malattia di VHL; de novo casi che producono mosaicismo genetico sono più
difficili da individuare perché le mutazioni non si trovano nelle cellule
bianche del sangue che vengono utilizzati per l'analisi genetica. [7] [15 ]
Trattamento
Non c'è modo di
invertire mutazioni VHL, ma il riconoscimento precoce e il trattamento di
specifiche manifestazioni di VHL può ridurre sensibilmente le complicanze e
migliorare la qualità della vita. Per questo motivo, gli individui con
malattia di VHL sono di solito sottoposti a screening di routine per angiomi
retinici, hemangioblastomas SNC, a cellule chiare carcinoma renale e
feocromocitomi. [16] hemangioblastomas
del SNC sono solitamente rimossi chirurgicamente se sono sintomatici. Fotocoagulazione e crioterapia di solito
vengono utilizzati per il trattamento di angiomi retinici sintomatici, anche se
i trattamenti anti-angiogenici possono anche essere un'opzione. Tumori
renali possono essere rimossi da un parziale nefrectomia o altre tecniche
come l'ablazione a radiofrequenza. [7]
Storia
.
L'oculista
tedesco Eugen von Hippel primo descrisse
angiomi nell'occhio nel 1904. [18]Arvid
Lindau descritto
i angiomi del cervelletto e della
colonna vertebrale nel 1927. [19] Il termine
malattia di von Hippel-Lindau stata la prima volta nel 1936, ma il suo utilizzo
divenne comune solo nel 1970.. [7]
La gente [modifica]
Alcuni discendenti
della famiglia McCoy (coinvolto nella faida Hatfield-McCoy di Appalachi si presume, USA)
per avere VHL. In un articolo apparso l'Associated Press, è stato
ipotizzato da un endocrinologo Vanderbilt University che l'ostilità alla base
della faida Hatfield-McCoy può essere stato in parte dovuto alle conseguenze
della malattia di von Hippel-Lindau. L'articolo suggerisce che la famiglia
McCoy è stato predisposto a malumore, perché molti di loro avevano un
feocromocitoma che ha prodotto in eccesso adrenalina e una tendenza verso gli
animi esplosivi. [20]
Nomenclatura
Altri nomi non comuni
sono: angiomatosi retine, familiare angiomatosi cerebello-retinica,
hemangioblastomatosis cerebelloretinal, malattie Hippel, sindrome di
Hippel-Lindau, HLS, VHL, Lindau malattia o angiomatosi retinocerebellar. [21] [22]
Sintomatologia
A seconda del genotipo del soggetto
sono rinvenibili quadri clinici differenti; in particolare la malattia è
suddivisibile in 2 tipi. Il tipo 1 con assenza di feocromocitoma e il tipo 2
che invece lo manifesta. Nel primo caso la proteina spesso è mutata, ma presente;
nel secondo caso invece è deleta. Inoltre il tipo 2 è diviso ulteriormente in 3
sottotipi: A, B e C (il primo con tumore a cellule renali, il secondo con anche
emangioblastoma e il terzo con solo feocromocitoma). Sono anche riscontrati
tumori endocrini (specie pancreatici), cisti renali (dovute all'edema da
neoangiogenesi) e altre manifestazioni neoplastiche.
Diagnosi
La ricerca delle alterazioni del
gene VHL deve essere guidata dalla clinica con cui la patologia si presenta e
dalla buona pratica clinica. (bisogna valutare l'utilità reale e la metodica
più adatta nel fare una consulenza genetica). In particolare nel caso in cui
sospetti una delezione dovrò fare gli esami adatti a trovarla, nel caso in cui
avrò invece una mutazione puntiforme userò delle metodiche adatte a cercare
queste ultime; fatto ciò nel secondo caso dovrò valutare se la mutazione
eventualmente trovata corrisponde a patologia; o tramite la consultazione di
database appositi oppure facendo delle prove indirette (cercò nei tumori del
soggetto anche la delezione dell'allele sano oltre a quello mutato
congenitamente, in modo da provare che il danno trovato è responsabile del
quadro clinico)
Terapia
Si
procede ad intervento chirurgico, che viene effettuato in particolari
condizioni:
Lesioni
spinali sintomatiche
Lesioni
cerebrali sintomatiche o in rapida evoluzione
Neoplasie
solide renali > 3 cm
Neoplasie
solide pancreatiche > 3 cm
Endolymphatic sac
tumors
affect about 10% of cases, and VHL should be considered in persons with
bilateral endolymphatic sac tumors.Tumori del sacco endolinfatico colpiscono circa il 10% dei casi, e
VHL dovrebbero essere considerati in soggetti con tumori bilaterali sacco endolinfatico.
Lesioni hemangioblastoma del
midollo spinale e del cervelletto in paziente con malattia di Von Hippel
Lindau
Atassie spino
cerebellari:
Tutti questi disturbi
esibiscono disfunzione pancerebellari gradualmente progressiva, di solito a partire
dall'infanzia, differenziati per altro coinvolgimento del sistema
nervoso. Questi disturbi sono stati precedentemente noto come autosomica
dominante atassie cerebellari. La prevalenza di SCA è stimata in circa 1-4
/ 100.000, ma può essere molto più elevata in alcune regioni a causa
dell'effetto fondatore. SCA sono tutti autosomica dominante.
SCA
Tipo
risultati
e commenti
Mutazione
SCA-1
(3-15%)
Saccades
ipèrmetro, saccadi lente, UMN
CAG
repeat, 6p
SCA2
(6-15%)
Saccades velocità diminuita, areflessia
Comune a Cuba.
CAG
repeat, 12q
SCA3
(MJD, 30-40%)
Gaze-evocato nistagmo, UMN,
saccadi lente.
Comune nelle Azzorre.
CAG
repeat, 14q
SCA-4
(17 famiglie)
areflessia
Cromosoma
16q
SCA-5
Pure
cerebellari
Cromosoma
11
SCA-6
Downbeating nistagmo, vertigine
posizionale
I sintomi possono comparire per la
prima volta il più tardi di 65 anni.
CAG
repeat, 19p (gene del canale del calcio)
SCA-7
Degenerazione
maculare, UMN, saccadi lente
CAG
repeat, 3p
SCA-8
Il
nistagmo orizzontale
CTG
repeat, 13q
SCA-9
SCA-10
(Zu et al, 5 famiglie)
atassia,
convulsioni, soprattutto in messicani
Cromosoma
22q collegato, ripetere pentanucleotide
SCA-11
15q
SCA-12
(rara, Ohearn et al)
Testa
e tremore alle mani, akinseia
5q
CAG
SCA-13
(raro)
Ritardo
mentale
19q
SCA-14
(raro)
Mioclono
19q
SCA-16
Testa
e la mano tremori
8q
DRPLA
Corea, convulsioni, trovano
principalmente in Giappone
Espansione
12p CAG
SCA
19, 22?
La
sindrome cerebellare Lieve, disartria
SCA
25
atassia
con neuropatia sensoriale, vomito e dolori gastrointestinali.
SCA1-3, SCA6-7m 12 e 16,
sono geneticamente instabili associati a trinucleotide ripetizioni
CAG. Trinucleotide ripetizioni sono anormali "nonsense" aree di
DNA umano, che tendono a diventare più grandi con il tempo. Nelle
generazioni successive, la dimensione della ripetizione CAG tende ad
ingrandirsi causando una diminuzione dell'età di insorgenza (chiamato
anticipazione). Altre malattie repeat CAG includono la malattia di Huntington, l'atrofia
dentatorubral-pallidoluysian, e atrofia muscolare spinale e
bulbare.Sorprendentemente, il CAG ripetizioni nel SCA1-3 si trovano su
cromosomi diversi. Altre malattie trinucleotide repeat includono distrofia
miotonica e sindrome dell'X fragile. In trinucleotide ripetizioni,
l'espansione può aumentare quando viene passato tra un genitore affetto e il
suo bambino malato - questo è chiamato anticipazione. Lo stato di
portatore di premutazione Fragile-X è anche associata con i risultati
cerebellari (Berry-Kravis et al, 2003). Questa mutazione ha una frequenza
di 1/250 di donne e uomini 1/813.
In SCA1 vi è l'atrofia
delle cellule di Purkinje, così come la perdita di molte proiezioni afferenti
alla corteccia cerebellare, atrofia dei percorsi dentatorubral, le colonne
dorsali e alcuni nuclei dei nervi cranici .. mappe SCA1 al cromosoma
6p. Ampiezza saccade è riferito aumentato in SCA1, con conseguente
ipermetria (Rivaud-Pechoux et al, 1998). In tribù dominante, Moseley et al
(1998) trovarono SCA1 nel 5,6%.
SCA2 è associata a
perdita marcata o il rallentamento dei movimenti oculari saccadici. C'è L’atrofia
olivopontocerebellare. SCA2 mappa al cromosoma 12q. SCA2 può essere
il più comune dei tipo CAG repeat autosomica dominante atassie
cerebellari. Velocità saccadico (rapido velocità movimento degli occhi) è
diminuita in SCA2 (Rivaud-Pechoux et al, 1998). In tribù dominante, Mosely
et al trovato SCA2 a 15,2%.
SCA3, che è dominante
ereditato, è conosciuta anche come malattia di Machado-Joseph, si veda anche la
voce OMIM.
SCA3 è caratterizzata
patologicamente da atrofia spinopontine, degenerazione dei nuclei dentati,
nuclei vestibolari, strutture extrapiramidali, nervi cranici motori, cellule
delle corna anteriori e posteriori radice gangliari, ma risparmiatori della
corteccia cerebellare. Clinicamente, è caratterizzata da atassia
cerebellare, segni piramidali e oftalmoplegia esterna progressiva. C'è
spesso retrazione palpebrale producendo una caratteristica espressione di
sguardi, chiamato occhi sporgenti. (Anche se questo è riportato in
letteratura, non abbiamo mai incontrato questo risultato nella nostra popolazione
clinica - e non pensare che questo è richiesto). Gaze nistagmo evocato è spesso presente
in SCA3 (Rivaud-Pechoux et al, 1998 ). Gambe senza riposo si verifica nel
45% (Schols, 1998). Fasciali e lingua fascicolazioni sono talvolta
presenti.
MJD è stato descritto
nelle famiglie di origine portoghese, ma è stato documentato in molte famiglie
non di origine portoghese. Diversi grandi studi hanno dimostrato che MJD
rappresenta circa il 84% di autosomica dominante SCA in portoghese, il 50% in
tedesco, e 11-29% in altre popolazioni di etnia non portoghesi. Il locus
MJD è stato mappato sul cromosoma 14q31.1, e la mutazione è stato dimostrato di
essere una espansione di una ripetizione CAG. (Soong et al,
1997). Atrofia del tronco cerebrale e del verme in MJD è strettamente
correlata sia con la dimensione della ripetizione CAG e l'età del paziente
(Onodera et al, 1998).
I test genetici è il
modo usuale che MJD viene diagnosticata. Sperimentazione clinica può
essere suggestiva, ma non vi è sovrapposizione tra molti altri SCA. VEMP
test (collo) può essere ridotto a MJD (SCA-3). Illustriamo un caso di questo qui.
SCA6 è un atassia
autosomica dominante associata a piccole espansioni di una ripetizione
trinucleotide (CAG) nella CACNL1A4 gene, che codifica per un canale del calcio
voltaggio-dipendenti. Zoghbi (1997) esamina la genetica di questo
disturbo.
I pazienti con SCA6
possono disporre di almeno tre differenti sindromi: atassia episodica, atassia
cerebellare più tronco encefalico o lungo degenerazione del tratto, o puro
atassia cerebellare. Canali del calcio sono identificati in Purkinje e
granuli neuroni. Clinicamente hanno un nistagmo grossolano
sguardo-evocato, nistagmo downbeat su sguardo laterale, e la scarsa
soppressione visiva (Gomez et al, 1997). SCA6 conti per circa il 30% delle
atassie dominanti in Giappone, e tra il 5-15% di atassia ereditaria dominante
negli Stati Uniti (Geshwind et al, 1997; Mosely et al, 1998). Studi di
imaging rivelano atrofia cerebellare con relativo risparmio del tronco
cerebrale. In Giappone, l'atassia è il sintomo iniziale più comune. I
pazienti con corsi prolungati mostrano posture distoniche, movimenti
involontari e anomalie nella riflessi tendinei (Ikeuchi et al,
1997). Takeichi et al (2000) ha riferito che mentre oculare esercizio regolare è diminuita, la
cancellazione vestibolare è normale. Questo può essere un risultato
caratteristico di questa condizione. Come accennato in precedenza, i
pazienti con canalopatie calcio comprese SCA-6 e EA2 hanno risposte oculari
carenti didell'otolite ingresso.
SCA7, anche ereditaria
dominante, è associata a retinopatia o cecità. E 'anche un disturbo CAG
repeat. Mosely et al (1998) trovarono SCA7 in circa il 5% delle atassie
ereditarie dominante.
SCA8 è stata descritta
nel 2000 da Ikeda e altri. Si tratta di un / CTG disturbo CAG
repeat. E 'caratterizzata da mancanza di coordinazione, disartria atassica,
alterata esercizio regolare, nistagmo orizzontale, e l'atrofia del verme
cerebellare e emisferi. Distrofia miotonica è un altro disturbo
ripetizione CTG. Entrambi mostrano anticipazione materna. Età media
di insorgenza è 53,8 anni.
Secondo Matsuura et al,
SCA 9 è riservato per i disturbi ancora essere descritti in letteratura, e
SCA10 (Zu et al, 1998), designa un'altra atassia autosomica dominante, con
crisi epilettiche occasionali.
SCA-10 è rara nelle
popolazioni diverse da messicani (Matsuura ed altri, 2002).
SCA-17 è un autosomica
dominante atassia cerebellare causate da espansione CAG repeat nel gene della
proteina TATA-box binding. Le caratteristiche cliniche comprendono
l'atassia, demenza, iperreflessia, manifestazioni parkinsonismo come la
bradicinesia, e disturbi riflessi posturali.
SCA-34 è autosomica
dominante atassia cerebellare a causa di una mutazione ELOVL4. Le
mutazioni di ELOV4 sono stati riportati in 2 parentele giapponesi e una
famiglia franco-canadese. Nella variante giapponese, il disordine è simile
PSP compreso il segno "hot cross bun" in qualche e hyperdensities
lineari pontine in altri. (Ozaki et al, 2015).
SCA-38 è dovuta a una
mutazione missense di ELOVL5. ELOV5 è coinvolto nella sintesi degli acidi
grassi. (Di Gregorio et al, 2014).
Ci sono molte altre
atassie che non sono inclusi nella nomenclatura "SCA #".
Grewal e altri (1998)
hanno descritto una nuova malattia spinocerebellare autosomica dominante negli
individui del patrimonio messicano-americano. Il quadro clinico comprendeva atassia
cerebellare, sguardo e rimbalzo nistagmo.
Matsuura ed altri (1999)
mapparono un autosomica dominante atassia spinocerebellare con crisi, anche in
una famiglia ispanica.
Swartz e altri (2003)
hanno descritto una nuova atassia autosomica ressive progressivo con atassia,
segni corticospinali, assonale sensitivo-neuropatia, e la rottura di fissazione
visiva da intrusioni saccadiche. Questo disturbo è stata associata a una
mutazione 1p36.
References
Berry-Kravis E and
others. Tremor and ataxia in fragile X premutation carriers: blinded
videotape study. Ann
Neurol 2003:53:616-623
Di Gregorio, E., et al. (2014). "ELOVL5
mutations cause spinocerebellar ataxia 38." Am J Hum Genet 95(2): 209-217.
Geshwind DH and
others. Spinocerebellar ataxia type 6: frequency of th emutaiton and
genotype-phenotype correlations. Neurology 1997:49:1247-1251
Gomez et
al. Spinocerebellar ataxia type 6: gaze-evoked and vertical
nystagmus, purkinje cell degeneration, and variable age of onset. Ann Neurol 1997:42:933-950
Ikeuchi T and
others. Spinocerebellar ataxia type 6: CAG repeat expansion... Ann Neurol 1997:42:879-884
Ikeda and
others. Molecular and clinical analyses of spinocerebellar ataxia
type 8 in Japan. Neurology
2000;54:950-965
Jen JC and
others. Spinocerebellar ataxia type 6 with positional vertigo and
acetazolamide responsive episodic ataxia. J. Neuro Neurosurg Psych
1998:65:565-568
Matsuura T, and
others. Mapping of the gene for a novel spinocerebellar ataxia with
pure cerebellar signs and epilepsy. Ann Neurol 1999:45:407-411
Matsuura T and
others. Spinocerebellar ataxia type 10 is rare in populations other
than mexicans. Neurology
58, 963, 2002
Mosely ML and
others. Indcidence of dominant
spinocerebellar and Freidreich triplet repeats among 361 ataxia families.Neurology 1998:51:1666-1671
O'Hearn E, Holmes
SE, Calvert PC, et al. Spinocerebellar ataxia type 12 (SCA12)
clinical and neuroradiologic features. Neurology 2001, 56:299-303
Onodera O, and
others. Progressive atrophy of cerebellum and brainstem as a function
of age and the size of the expanded CAG repeats in MJD1 gene in
Machado-Joseph Disease. Ann
Neurol 1998:43:288-296
Ozaki, K., et
al. (2015). "A Novel Mutation in ELOVL4 Leading to
Spinocerebellar Ataxia (SCA) With the Hot Cross Bun Sign but Lacking Erythrokeratodermia:
A Broadened Spectrum of SCA34." JAMA Neurol 72(7): 797-805.
Rivaud-Pechoux S and
others. Eye movement abnormalities correlated with genotype in
autosomal dominant cerebellar ataxia type I. Ann Neurol 1998:43:297-302
Schwartz BE and others. Pathogenesis
of clinical signs in recessive ataxia with saccadic intrusions. Ann Neurol 2003: 54:824-828
Soong BW, Cheng CH,
Liu RS, Shan DE. Machado-Joseph disease: clinical, molecular, and
metabolic characterization in Chinese kindreds. Ann Neurol 1997:41:446-452
Subramony SH, Filla
A. Autosomal dominant spinocerebellar ataxias and infinitum. Neurology 2001:56:287-289
Takeichi and
others. Dissociation of smooth pursuit and vestibulo-ocular reflex
cancellation in SCA-6. Neurology
2000:54:860-866
Wiest et
al. Otolith functiobn in cerebellar ataxia due to mutations in the
calcium channel gene CACNA1A Brain (2001) 2407-2416
Zoghbi HY. CAG
repeats in SCA6. Anticipating
new clues. Neurology 1997:49:1196-1199
Zu L and
others. Mapping of a new autosomal dominant spinocerebellar ataxia
(SCA8) to chromosome 22. Am
J. Hum Genet 1998:63(Suppl):A347
L’atassia
spino-cerebellare di tipo 6 (SCA6)
The
spinocerebellar ataxia type 6 (SCA6)
La SCA6 è il sottotipo più comune delle ADCA di tipo III e costituisce circa un
terzo di tutte le atassie cerebello-spinali. Esiste una notevole variabilità
dei valori di prevalenza nelle diverse aree geografiche con oscillazioni nel
range tra 0,02 a 0,31 per 100.000 abitanti.
Si tratta di una atassia cerebellare ereditaria di tipo autosomico dominante ad
insorgenza tardiva (media 4.5 anni con comparsa oltre i 50 anni nel 60%
dei casi), assai lentamente progressiva (ed abitualmente “non fatale”),
caratterizzata principalmente da segni di disfunzione cerebellare ed oculomotoria
(diplopia nel 50% dei casi) che solo occasionalmente e per lo più tardivamente
si associa ad altri sintomi di tipo piramidale, extrapiramidale o di lieve
deterioramento cognitivo. La maggior parte dei pazienti presenta disturbi della
deambulazione di tipo atassico cerebellare ed espisodi di disequilibrio (in
circa il 90% dei casi) o di vertigine episodica (60%) come sintomo di esordio
mentre, molto più raramente, i primi sintomi della malattia possono essere
diplopia, disartria, tremori o sensazioni di stordimento nella rapida rotazione
della testa. Nei familiari di questi pazienti si possono riscontrare soggetti
affetti da “emicrania emiplegica”.
Tra i sintomi e segni di interesse otoneurologico sono molto frequenti i
sintomi correlati alla difficoltà di fissare oggetti in movimento e anche il
nistagmo orizzontale evocato dallo sguardo (70-100%) o quello verticale
(65-83%) anche in fasi precoci della malattia (Yabe et al., 2003). Sono
state descritte anche varie altre anomalie dei movimenti oculari, incluso il
nistagmo di rimbalzo ed il periodico alternante ( Wai-Man et al., 2009). Sono
stati anche descritti (Jen et al., 1998) alcuni casi di SCA6
caratterizzati da vertigine posizionale e crisi di atassia episodica responsiva
all’acetazolamide.
La SCA6 è dovuta ad una piccola espansione della tripletta CAG nel gene CAGNA
lA che codifica la subunità alfa i del canale cerebrale del calcio tipo P/Q
voltaggio-dipendente localizzato nel cromosoma 19p. I soggetti affetti da SCA6
hanno da 20 a 33 ripetizioni della tripletta CAG.
E noto che alterazioni dello stesso gene sono la causa di altre due malattie
ereditarie e cioè dell’Emicrania Erniplegica Familiare e 1’ atassia episodica
di tipo 2 ed esistono elementi cImici ed epidemiologici che fanno considerare
queste due malattie, unitamente alla SCA6, come affezioni alleliche con
sovrapposizioni fenotipiche da mutazione del gene CACNA1A (Jen etal., 1998).
L'atassia di Friedreich
(FRDA, FA) e ancora un altro esempio di malattia causata dall'espansione
ripetuta di un trinucleotide. FRDA e causata dall'espansione di una tripletta
di GAA localizzata all'interno del primo introne del gene della frataxina. C'e
una correlazione chiara tra dimensione della ripetizione espansa e la gravita
del fenotipo in FRDA. La frataxina e una proteina mitocondriale che ha un ruolo
nell'omeostasi del ferro. La deficienza di frataxina da luogo ad accumulo
mitocondriale di ferro, a difetti a carico di enzimi mitocondriali specifici,
ad aumentata sensibilità agli stress ossidativi, ed infine alla morte cellulare
mediata da radicali liberi. FRDA e considerata una nuova malattia mitocondriale
codificata nel nucleo. Strategie moderne che usano scavengers dei radicali
liberi offrono la speranza almeno di rallentare la progressione della
cardiopatia in questi pazienti. Modelli murini di FRDA sono stati sviluppati
mediante gene targeting e dovrebbero offrire sistemi in cui testare potenziali
terapie.
L'atassia di Friedreich
fu riportata per la prima volta nel 1863 da Nicholaus Friedreich ad Heidelberg,
Germania. Egli descrisse i reperti essenziali della malattia: atrofia
degenerativa dei cordoni posteriori del midollo spinale che conducono ad
atassia progressiva, perdita di sensibilità, e debolezza muscolare, spesso
associata con scoliosi, deformità del piede, e cardiopatia. Anche se fu
riportato inizialmente più di 150 anni fa, lo spettro clinico completo di FRDA
e le caratteristiche che hanno distinto questa malattia dalle altre sindromi
atassiche sono stato controversi. Il Quebec Collaborative Group nel 1976 e
Harding nel 1981 definirono i criteri clinici essenziali per la diagnosi di
FRDA. Con l'applicazione di metodi di genetica molecolare, sono stati
identificati i loci e le mutazioni per molte atassie ereditarie. Il gene di FRDA
fu localizzato nel cromosoma 9q, e la mutazione più comune fu definita come
un'espansione instabile di una sequenza ripetuta della tripletta nucleotidica
GAA. Fin da questa scoperta, la diagnosi molecolare di casi atipici e divenuta
possibile e lo spettro fenotipico di FRDA e stato ampliato. FRDA e l'atassia
ereditaria piu comune. E una malattia neurodegenerativa autosomica recessiva
con una prevalenza stimata di 1:50000-1:29000. L'incidenza e molto piu bassa
negli asiatici e nei soggetti di discendenza africana. La percentuale dei
portatori e stata stimata a 1:120-1:60 (Alper G and Narayanan V. 2003.
Friedreich's Ataxia. Pediatr Neurol 28: 335-41).
L'atassia progressiva ed
incessante e la caratteristica principale di questa malattia. Piu comunemente,
comincia con goffaggine nella marcia. L'esordio di solito e verso la puberta,
ma puo variare da 2-3 anni di eta a oltre 25 anni di eta. Secondo Harding le
caratteristiche cliniche essenziali sono (1) eredita autosomica recessiva, (2)
esordio prima dei 25 anni di eta, (3) progressiva atassia degli arti e della
marcia, (4) riflessi tendinei assenti alle estremita inferiori, (5) evidenza
elettrofisiologica di neuropatia assonale sensitiva seguita da (entro 5 anni
dall'esordio), (6) disartria, (7) areflessia in tutti i quattro arti, (8)
perdita distale della sensibilita di posizione e pallestesica, (9) risposta
plantare in estensione, e (10) debolezza piramidale delle gambe. La perdita
della deambulazione avviene mediamente 15.5 ? 7.4 anni dopo l'esordio della
malattia. La cardiomiopatia e la causa piu comune di morte. Si considera che i
pazienti che esibiscono tutte queste caratteristiche cliniche elencate da
Harding abbiano la "tipica" o "classica" forma della
malattia. Come con l'eta di esordio, vi e una grande variabilita nelle altre
caratteristiche cliniche, tra cui la velocita di progressione, la gravita, e
l'estensione del coinvolgimento da parte della malattia. I pazienti possono
essere confinati su una sedia a rotelle nella prima adolescenza o possono
essere ancora deambulanti alla fine della quarta decade di vita. Le
complicazioni cardiache possono essere gravi abbastanza per provocare la morte
precocemente o possono essere minime o assenti. Altri reperti neurologici
variabili includono anormalita oculomotorie, nistagmo, atrofia ottica (25%), e
perdita d'udito neurosensoriale (20%). L'anomalia del movimento oculare piu
comune e l'instabilita nello sguardo fisso. Non si osserva oftalmoparesi. Piu
di meta dei pazienti manifesta scoliosi progressiva, e circa meta manifesta
piede cavo, piede equinovaro e dita dei piedi ad artiglio. L'amiotrofia dei
piccoli muscoli della mano e dei muscoli distali della gamba e del piede e
comune. Circa il 10% dei pazienti FRDA sono diabetici, e la maggior parte dei pazienti
diabetici richiede terapia insulinica. La cardiomiopatia e evidente in circa i
due terzi dei pazienti con FRDA ed e primariamente una cardiomiopatia
ipertrofica concentrica simmetrica, anche se alcuni pazienti esibiscono
ipertrofia settale asimmetrica. L'elettrocardiogramma illustra ampie inversioni
dell'onda T e segni di ipertrofia ventricolare. Dopo la scoperta del difetto
molecolare in FRDA, i pazienti con atassia autosomica recessiva o sporadica
furono esaminati per rivaluare lo spettro fenotipico. La vasta maggioranza
(93-96%) dei pazienti con caratteristiche tipiche di FRDA e omozigote per
l'espansione di GAA; inoltre, anche alcuni pazienti che non presentano FRDA
classica o tipica (quelli che non soddisfano i criteri di Harding) erano omozigoti
per la ripetizione espansa della tripletta nucleotidica. Il fenotipo di FRDA e
stato ampliato per includere le varianti seguenti che sono considerate FRDA
atipica:
FRDA ad esordio tardivo
Eta
di esordio >20 anni fu riscontrata in 19 di 114 pazienti (17%) con un
fenotipo di FRDA. Non c'era evidenza di eterogeneita genetica. Un confronto dei
loro reperti clinici e di laboratorio con quelli dei pazienti che avevano
sviluppato FRDA all'eta tipica rivelo solamente una minor incidenza di
anormalita scheletriche (piede cavo e scoliosi). La progressione della malattia
come indicata dagli anni tra l'esordio ed il confinamento alla sedia a rotelle
era piu lenta nei pazienti con FRDA ad esordio tardivo.
FRDA con conservazione dei riflessi
Palau
et al. dimostrarono un legame con il locus FRDA in otto pazienti da sei
famiglie. Tutti i pazienti soddisfacevano ai criteri diagnostici di Harding, a
parte l'assenza di scosse a ginocchio e caviglia. Comunque, manifestavano tutti
neuropatia assonale sensitiva alle prove neurofisiologiche. Tutti i pazienti
dimostrarono anomalie all'elettrocardiogramma. L'evidenza di cardiomiopatia
ipertrofica era presente all'ecocardiogramma in tutti tranne uno. C'era
coesistenza di fenotipo FRDA tipico in alcune famiglie. Un altro studio descrisse
una famiglia con un link al locus del cromosoma 9; un membro esibi FRDA tipica,
ed un altro manifesto una sindrome atassica simile con riflessi conservati. Il
paziente con riflessi tendinei conservati non dimostro evidenza
elettrofisiologica di neuropatia assonale afferente grave usuale in FRDA.
Tipo Acadiano (Forma Louisiana)
Il tipo Acadiano
osservato in una specifica popolazione di origine francese che vive nel nord
America fu distinto da FRDA tipica dal suo decorso piu mite e la minore
incidenza di cardiomiopatia. E stata dimostrata omogeneita di linkage tra la
forma Acadiana e la classica FRDA (Alper G and Narayanan V. 2003. Friedreich's
Ataxia. Pediatr Neurol 28: 335-41).
L'esordio
di FA e precoce, e l'atassia alla deambulazione e il sintomo usuale di
presentazione. Tipicamente entrambi gli arti inferiori sono colpiti in modo
uguale. Alcuni pazienti possono avere inizialmente emiatassia prima che i
sintomi divengano generalizzati. In certi casi, l'atassia comincia
improvvisamente dopo una malattia febbrile nella quale l'atassia di un arto
inferiore precede quella dell'altro. L'atassia della andatura si manifesta come
marcia progressivamente lenta e goffa, che spesso comincia dopo che si e
sviluppato una normale capacita di deambulazione. L'atassia puo essere
associata con difficolta alla stazione eretta e nella corsa. L'atassia e di
tipo sia sensoriale sia cerebellare. Questa combinazione e stata etichettata
come marcia tabetocerebellare. Vi sono opinioni contrastanti se predominino le
caratteristiche sensoriali o le cerebellari. Le caratteristiche cerebellari
dell'atassia nella marcia di FA includono una andatura a base allargata con
cambiamento continuo di posizione per mantenere l'equilibrio. Sedersi e alzarsi
in piedi sono manovre associate ad esitazione. L'atassia sensoriale che e il
risultato di una perdita del senso di posizione articolare contribuisce alla
stazione e all'andatura a base allargata, ma e presente anche una marcia
steppante, caratterizzata da colpi disuguali ed irregolari del pavimento con la
pianta dei piedi. Tentativi di correzione di ogni squilibrio danno tipicamente
luogo a movimenti improvvisi e violenti. Al progredire della malattia,
l'atassia colpisce il tronco, le gambe, e le braccia. Quando le braccia divengono
macroscopicamente atassiche, si possono sviluppare sia tremori d'azione sia
tremori intenzionali. Può comparire esitazione del tronco. I muscoli facciali,
buccali, e delle braccia possono essere colti da tremori e talvolta essere
interessati da movimenti coreiformi. Il paziente può esperimentare facile
affaticabilità.
Pazienti
con FA avanzata possono avere intensa debolezza distale delle gambe e dei
piedi, anche se una debolezza significativa delle braccia e rara prima che il
paziente sia costretto in un letto. Infine, il paziente non e capace di
camminare a causa della debolezza progressiva e dell'atassia, e diventa cosi
prima legato ad una sedia a rotelle e da ultimo costretto nel letto.
Con
il procedere della malattia, compaiono disartria e disfagia. L'eloquio diventa
via via indistinto, lento, e, alla fine, incomprensibile. I pazienti possono
esperimentare lieve indebolimento dei muscoli facciali con associata debolezza
alla deglutizione. L'incapacità a coordinare respiro, parola, deglutizione e
risata puo far si che il paziente quasi si soffochi mentre parla (Ciliberti E
and Galvez-Jimenez N. 2002. Friedreich Ataxia. eMedicine).
FA classica e il
risultato di una mutazione genica nella regione centromerica del cromosoma 9
(9q13-21.1) nel sito del gene che codifica per la frataxina, una proteina di
210 aminoacidi. Questa mutazione e caratterizzata da un numero eccessivo di
ripetizioni della tripletta nucleotidica GAA (guanina-adenina-adenina) nel
primo introne del gene che codifica per la frataxina. E l'unica malattia nota
che sia il risultato di una ripetizione del trinucleotide GAA. Questa
espansione altera l'espressione del gene, riducendo la sintesi della frataxina.
Si pensa che la ripetizione espansa di GAA dia luogo a deficit di frataxina
interferendo con la trascrizione del gene attraverso lo sviluppo di una
struttura elicoidale instabile. Maggiore il numero di ripetizioni, maggiore la
riduzione in espressione di frataxina. In un normale cromosoma, questa sequenza
trinucleotidica e ripetuta fino a 50 volte. In pazienti con FA, questa sequenza
e ripetuta almeno 200 volte e spesso più di 1000 volte. La variabilità nella
presentazione clinica di FA può essere spiegata dall'estensione dell'espansione
di questo trinucleotide ripetuto. L’età di esordio della malattia, la sua
gravita, la velocita di progressione e l'estensione del coinvolgimento
neurologico variano col numero di sequenze ripetitive di GAA. Inoltre, la
frequenza delle risposte dell'estensore plantare, della cardiomiopatia, della
debolezza e dell'affaticabilità delle gambe, della sclerosi e del piede cavo
aumentano con la grandezza dell'espansione di GAA. Le maggiori espansioni di
GAA correlano con una più precoce età di esordio e tempi più corti alla perdita
della deambulazione. In un recente studio condotto da Durr et al., la grande
maggioranza dei pazienti con FA (94%) era omozigote per il trinucleotide GAA
(l'espansione di GAA era presente su ambo gli alleli del gene della frataxina).
Il restante 6% era costituito da eterozigoti per l'espansione di GAA ed una
mutazione puntiforme della frataxina (uno allele aveva un'espansione di GAA e
l'altro aveva una mutazione puntiforme senza espansione). Non sono mai stati
descritti pazienti omozigoti per una mutazione puntiforme. Le mutazioni
puntiformi non solo riducono i livelli di frataxina, ma sono anche responsabili
della formazione di una proteina anormale. Esse rappresentano anche un'altra
fonte di variabilità nella presentazione clinica di FA. Diciassette diverse
mutazioni puntiformi sono state finora descritte in FA. Tra l'1% e il 5% delle
mutazioni puntiformi e rappresentato da cambiamenti di una singola base nella
sequenza del gene di FA tale da provocare mutazioni missense, nonsense o
splicing. I pazienti con mutazioni missense presentano sintomi lievi o gravi,
mentre mutazioni di splicing, nonsense e acrico del codone di iniziazione,
associate alla sintesi di una frataxina non funzionante, danno luogo ad un
fenotipo grave. Mutazioni puntiformi del gene della frataxina che coinvolgono
il terminale aminico tipicamente si manifestano con un decorso piu benigno che
quelle che coinvolgono il terminale carbossilico. Le tre mutazioni puntiformi
più comuni sono la mutazione II54F nell'Italia meridionale, la mutazione
ATG>ATT del codon iniziale e la mutazione G130V. I pazienti con quest'ultima
mutazione tendono ad avere una piu lenta progressione di malattia, riflessi
patellari attivi, e minima disartria. Le cellule e i tessuti dell'organismo
sono sensibili in modo diverso al deficit di frataxina. Le cellule che
normalmente richiedono e producono le maggiori quantità di frataxina tendono ad
essere le piu colpite da FA. Per esempio, i neuroni sensitivi presenti nel
ganglio delle radici dorsali responsabili della percezione della posizione
nello spazio esprimono marcatamente il gene della frataxina e sono colpiti
pesantemente in FA. Anche le fibre muscolari miocardiche richiedono quantità di
frataxina maggiori in confronto con altri tessuti e sono pesantemente colpite
in FA. Un certo numero di esperimenti ha confermato la localizzazione
subcellulare mitocondriale della frataxina nei mammiferi. E stato dimostrato
che la frataxina e essenziale per la normale funzione mitocondriale, sia per la
fosforilazione ossidativa sia per l'omeostasi del ferro. Esiste forte evidenza
che il deficit di frataxina da luogo ad accumulo di ferro nei mitocondri di
cellule affette in linee cellulari in coltura. Apparentemente, la velocita
dell'esporto mitocondriale e ridotta. Cuori di pazienti con FA hanno rivelato
depositi simil-ferrosi mitocondriali che non sono presenti in cuori sani.
L'accumulo eccessivo di ferro nei mitocondri riguarda i livelli citosolici di
ferro. L'eccesso di ferro intracellulare stimola un'aumentata produzione di
radicali liberi ed induce danno mitocondriale. L'eccesso di ferro inattiva gli
enzimi mitocondriali essenziali per la produzione di adenosin-trifosfato (ATP).
Ne consegue la morte cellulare, particolarmente dei neuroni del midollo spinale
e del sistema nervoso periferico. Un modello murino di FA e in via di sviluppo
per confermare l'evidenza di questo processo in modelli animali viventi
(Ciliberti E and Galvez-Jimenez N. 2002. Friedreich Ataxia. eMedicine).
La
consulenza genetica e disponibile per la diagnosi prenatale per genitori con un
figlio affetto. Lo screening di popolazione per i portatori del gene difettoso
non e utile. Un test specifico per l'espansione della ripetizione
trinucleotidica e commercialmente disponibile negli Stati Uniti e dovrebbe
essere effettuato in tutti i casi sospetti di FA. Non esiste alcuna evidenza di
anomalie del CSF nei pazienti con FA.
La risonanza magnetica (MRI) e lo metodica di scelta nella valutazione dei
cambiamenti in senso atrofico visti in FA. L'MRI cerebrale e midollare nei
pazienti con FA mostra tipicamente atrofia del midollo spinale cervicale con
minima evidenza di atrofia cerebellare.
L'ecocardiogramma
rivela ipertrofia ventricolare simmetrica, concentrica, anche se alcuni hanno
ipertrofia settale asimmetrica.
Circa
il 65% dei pazienti con FA ha reperti anomali all'ECG: i piu comuni sono
inversione dell'onda T, particolarmente nelle derivazioni standard inferiori e
toraciche laterali, ed ipertrofia ventricolare.
La
velocita di conduzione nervosa (NCV) in FA di solito e normale o solo
lievemente ridotta. I potenziali d'azione neurosensoriali (SNAP) sono assenti
in piu del 90% dei pazienti con FA. Il rimanente 10% mostra SNAP di ampiezza
ridotta.
Le risposte evocate uditive del tronco cerebrale sono tipicamente anormali in
FA, con assenza evidente di ondedi tipo III e IV e conservazione delle onde di
tipo I. Questo e suggestivo di coinvolgimento delle vie uditive centrali.
I potenziali
evocati visivi sono anormali in due terzi dei pazienti con FA. Si registrano
latenza assente o ritardata ed ampiezza ridotta dell'onda di p100.
I
potenziali evocati somatosensoriali (SSEP) rivelano potenziali ritardati e
dispersi nella corteccia sensitiva, cosi come anomala conduzione motoria
centrale (Ciliberti E and Galvez-Jimenez N. 2002. Friedreich Ataxia.
eMedicine).
Anche
i reperti neuropatologici sono specifici per FRDA e rendono differente questo
disturbo dalle altre atassie ereditarie. La perdita di grandi neuroni primari
nei gangli delle radici dorsali e considerato un reperto precoce. Questo
processo e seguito da degenerazione dei cordoni posteriori, dei tratti spino
cerebellari, e dei tratti motori corticospinali del midollo spinale e dell'atrofia
delle grandi fibre sensitive nei nervi periferici. Il cordone di Clarke e reso
atrofico, mentre i motoneuroni delle corna anteriori sono risparmiati. Nel
tronco encefalico i nuclei gracile e cuneato, dove terminano i cordoni
posteriori del midollo spinale, sono gravemente atrofici, indicando
degenerazione transsinaptica. I tratti lunghi, particolarmente le fibre del
tratto corticospinale, sono severamente colpiti nelle parti distali, suggerendo
una degenerazione retrograda. Le radici in entrata dei nervi cranici sensitivi
sono colpite, come lo sono i sistemi uditivo e vestibolare, mentre i nuclei dei
nervi cranici sono normali. Nel cervelletto la corteccia e risparmiata fino
alle fasi piu tardive del decorso della malattia, quando si puo osservare
perdita di cellule di Purkinje, ma i nuclei dentati sono severamente
atrofizzati. E stata evidenziata anche perdita di cellule piramidali (di Betz)
nella corteccia cerebrale, ma in misura minore. Nel sistema nervoso periferico,
la neuropatia sensitiva assonale e il reperto caratteristico. E stata osservata
la perdita maggiore di grosse fibre mieliniche. Questa perdita selettiva si
verifica anche nelle cellule gangliari delle radici dorsali. La natura dei
cambiamenti nel sistema nervoso periferico non e stata ancora ben compresa.
Ricercatori precedenti conclusero che c'e un difetto di maturazione negli
specifici neuroni che rimangono ipotrofici e di conseguenza lentamente vanno
incontro a morte retrograda. Venne suggerito un difetto nella sintesi e nel mantenimento
delle proteine strutturali (Alper G and Narayanan V. 2003. Friedreich's Ataxia.
Pediatr Neurol 28: 335-41).
FA e caratterizzata da
eredita autosomica recessiva. In famiglie con un figlio affetto, il rischio
susseguente di un altro figlio affetto e 25%. Come nella maggior parte delle
malattie recessive, il rischio di sviluppare FA e massimo se la nascita
consegue all'unione tra consanguinei. In nord America ed Europa, comunque, i
casi sono per lo piu sporadici e si manifestano in famiglie di non
consanguinei. Il rischio complessivo di un paziente con FA di avere un figlio
con la stessa condizione e approssimativamente 1 su 200, a meno che vi sia consanguineità.
Se quello stesso paziente ha un partner che si scopre essere un portatore di
FA, allora il rischio diventa di 1 su 2. I figli del fratello non affetto di un
paziente con FA e di una sposa non consanguinea hanno un rischio di 1:1000 di sviluppare
FA. Il test per sapere se si e portatori e disponibile per i parenti di
pazienti affetti ed i loro partner. Comunque, anche il piccolo rischio di una
mutazione puntiforme deve essere preso in considerazione e registrato in una
consulenza genetica. E disponibile anche un test di mutazione diretta per la
diagnosi prenatale (Ciliberti E and Galvez-Jimenez N. 2002. Friedreich Ataxia.
eMedicine).
Lo stress ossidativo
sembra giocare un ruolo chiave nella patogenesi di FRDA, sia che l'accumulo
mitocondriale di ferro sia primitivo o secondario. I primi trial terapeutici erano
con chelanti del ferro come la desferrioxamina che riduce il ferro
intracellulare, ma la cui capacita di rimuovere il ferro mitocondriale e
ignota. Nei pazienti con FRDA sono state dimostrate sideremia e ferritina
normali. Inoltre, studi in vitro hanno rivelato che la desferrioxamina causa
riduzione dell'attività aconitasica in presenza di ferro ridotto. Gli
antiossidanti sono considerati potenziali agenti terapeutici per ridurre i
radicali liberi. Rustin et al. hanno usato l'antiossidante idebenone, un
analogo a corta catena del coenzima Q10, in 40 pazienti FRDA. Tutti i pazienti
manifestavano cardiomiopatia ipertrofica. L'idebenone protegge in vitro le
proteine contenenti gruppi sulfo-ferrosi dai radicali derivati dalla chimica di
Fenton. Ai pazienti venne somministrato idebenone per os (5mg/kg/day in tre
dosi refratte) per un periodo di 6 mesi dopo il quale l'ecocardiogramma rivelo
più del 20% di riduzione della massa ventricolare sinistra in circa la meta dei
pazienti e nessun cambio significativo nell'altra meta. Questi risultati sono
promettenti per l'efficacia dell'idebenone nel controllo della cardiomiopatia
in FRDA. Un studio pilota ha valutato l'effetto del trattamento con coenzima Q
e vitamina E in pazienti FRDA. La spettroscopia magnetica con fosforo (31P-MRS)
e stata usata per svelare il metabolismo energetico prima e dopo 3 e 6 mesi
dall'inizio della terapia e ha rivelato miglioramento nella bioenergetica in
vivo del muscolo cardiaco e scheletrico. Il follow-up degli stessi pazienti per
24 mesi ha dimostrato un miglioramento sostenuto del metabolismo energetico del
muscolo cardiaco e scheletrico associato con assenza di progressione dei segni
sia neurologici sia ecocardiografici. Piu ampi trial controllati sono necessari
per confermare questi risultati incoraggianti e per determinare se lo stato
neurologico possa essere migliorato (Alper G and Narayanan V. 2003. Friedreich's Ataxia.
Pediatr Neurol 28: 335-41).
Atassia spinocerebellare
di tipo 6 (SCA6) è caratterizzata da insorgenza nell'età adulta, lentamente
progressiva atassia cerebellare, disartria e nistagmo. L'età media di
insorgenza è 43 a 52 anni. I sintomi iniziali sono instabilità
dell'andatura, inciampando, e lo squilibrio (in ~ 90%) e disartria (in ~
10%). Alla fine tutte le persone hanno andatura atassia, degli arti
superiori mancanza di coordinazione, tremore intenzionale, e disartria. La
disfagia e soffocamento sono comuni. Disturbi visivi possono derivare da
diplopia, difficoltà fissarsi su oggetti in movimento, orizzontale nistagmo
sguardo-evocato, e nistagmo verticale. Iperreflessia e risposte plantari
estensori si verificano in circa il 40% -50%. Segni dei gangli della base,
tra cui distonia e blefarospasmo, si verificano in fino al 25%. Mentazione
viene generalmente conservato.
Diagnosi / testing.
CACNA1A è l'unico gene in cui mutazione è noto per
provocare SCA6. La diagnosi di SCA6 basa sull'uso dei test genetici
molecolari per rilevare un anomalo CAG trinucleotide repeat espansione CACNA1A. Gli
individui affetti hanno da 20 a 33 ripetizioni CAG. Test molecolari rivela
un'espansione in più del 99% dei colpitiindividui.
Gestione.
Trattamento delle
manifestazioni: Acetazolamide per eliminare gli episodi di
atassia; soppressori vestibolari per ridurre vertigini e / o
osscilopsia; consultazione oculistica per la gestione di rifrazione o
chirurgico di diplopia;clonopin per disturbi del sonno REM; modifiche a
casa per la sicurezza e la convenienza; canne, bastoni da passeggio, e gli
escursionisti per evitare di cadere; terapia fisica per massimizzare il
risarcimento e la forza; logopedia e la comunicazione dispositivi per
disartria; ponderata posate e ganci medicazione; Video esophagrams di
individuare i comportamenti più sicuri e consistenza del cibo meno probabilità
di innescare l'aspirazione; alimentando la valutazione quando la disfagia
diventa fastidioso; controllo del peso, come l'obesità aggrava deambulazione
e mobilità difficoltà;CPAP per l'apnea del sonno.
Sorveglianza: Valutazione
annuale o semestrale da un neurologo; capacità di guida deve essere
valutata da professionisti periodicamente.
Agenti / circostanze da
evitare: sedativi
ipnotici (etanolo o alcuni farmaci) che aumentano incoordinazione.
Altro: farmaci Tremor
controllo di solito non sono efficaci nel ridurre i tremori cerebellari.
Consulenza genetica.
SCA6 è ereditata
come autosomica dominante maniera. Prole
di affetti individui hanno
una probabilità del 50% di ereditare la CACNA1Amutazione. Test prenatale è
possibile per le gravidanze ad alto rischio se la diagnosi è stata confermata
in un membro della famiglia; tuttavia, le richieste di diagnosi prenatale di malattie (in
genere) ad insorgenza nell'età adulta non sono comuni.
Atassia spinocerebellare
di tipo 6 (SCA6) è sospettata nei soggetti con insorgenza nell'età adulta,
lentamente progressiva atassia cerebellare, disartria e nistagmo. Perché
le manifestazioni fenotipiche di SCA6 non sono specifici, la diagnosi di SCA6
si basa su test di genetica
molecolare.
Molecolare Test
genetici
Gene.CACNA1A è
l'unico gene in cui mutazione è noto per
provocare SCA6.
Dimensioni allele. Una ripetizione
CAG polimorfici in esone 47 di CACNA1A è
instabile e si espande in individui con SCA6. Di seguito sono allele dimensioni
per CACNA1A:
Gli alleli di importanza
discutibile. 19
ripetizioni CAG. Il significato clinico di alleli con 19 CAG
ripetizioni non è chiara perché alleli di queste dimensioni sono state
documentate in quanto segue:
Espansione meiotica di una
ripetizione 19-CAG allele nella nota gamma
patogenicità [Mariotti et al 2001,Shimazaki et al 2001]. In questo caso, l'allele
è considerato un "allele intermedio" o "allele
normale mutabile" (cioè,
non è malattia che causa ma predispone all'espansione nell'intervallo
anormale).
Complete penetranza alleli. 20-33 CAG ripete [Jodice et al 1997,Yabe et al 1998]. Individui asintomatici
recanti un ampliamento della (CAG) 20 o superiore
sono tenuti a sviluppare sintomi in qualche momento della loro
vita. Le causano malattie più comuni allele ha 22 CAG ripetizioni.
La
capacità del metodo utilizzato per rilevare una mutazione che è presente
nella indicata gene
Strategia dei saggi
Per confermare /
stabilire la diagnosi in un probando,test di genetica molecolare di CACNA1A deve
essere eseguita per rilevare l'espansione CAG-repeat CACNA1A. Il
test può essere eseguito come un singolo gene test o come parte
di un pannello multi-gene che valuta per una varietà di condizioni atassia
ereditarie.
Test predittivi per asintomatici
membri adulti della famiglia a rischio richiede la preventiva individuazione
della diagnosi nella famiglia. Vedere correlati genetici
Problemi Counseling.
La diagnosi prenatale e
diagnosi genetica preimpianto (PGD) per gravidanze a rischio richiedono previa conferma della
diagnosi nella famiglia.
Geneticamente
Correlate (alleliche) Disturbi
Diversi altri disturbi
sono causati da mutazioni in CACNA1A.
Le atassie ereditarie
dominante possono
essere causati da mutazioni missense CACNA1A, tra cui
p.Gly293Arg o p.Arg1664Gln. Questi disturbi sono simili a SCA6, ma può
avere un quadro clinico più grave [Yue et al 1997,Tonelli et al 2006].
Episodica atassia tipo
2 (EA2)
è causata da
mutazioni CACNA1A che predicono proteina truncation, anormalesplicing, o mutazioni missense. Si
inizia in genere durante l'infanzia o nella prima adolescenza ed è
caratterizzata da attacchi di atassia, vertigini e nausea che durano ore o
giorni. Gli attacchi possono essere associati con disartria, diplopia,
tinnito, distonia, emiplegia, e mal di testa. Tra gli attacchi, gli
individui possono inizialmente essere normale, ma alla fine di sviluppare i
risultati interictali che possono includere nistagmo e atassia. Dopo anni
di atassia episodica, una condizione di atassia interictale indistinguibile da
SCA6 può sviluppare [Baloh et al 1997]. La trasmissione èautosomica dominante.
Pure FHM (che si trova nel 80%
dei colpiti famiglie), in cui l'esame
interictale è normale in tutti i membri della famiglia
FHM con sintomi cerebellari
permanenti (presenti nel 20% dei colpiti famiglie), in cui alcuni
membri della famiglia mostrano nistagmo e / o atassia interictale
Circa il 50% delle
famiglie con FHM, compresi tutti quelli con sintomi cerebellari permanenti,
hanno mutazioni missense in CACNA1A[Battistini ed altri 1999,Ducros ed altri 1999,Amico et al 1999]. FHM è
caratterizzata da un alone di emiplegia che è sempre associato ad almeno un
altro sintomo aura (emianopsie, deficit hemisensory, afasia).L'aura è seguito
da un moderato a grave cefalea. Il fenotipo comprende coma e
convulsioni [Ducros et al 2001], che può essere
attivato da trauma cranico lieve o angiografia. Edema cerebrale ritardato
è visto soprattutto in bambini e adolescenti che subiscono un trauma cranico
minore, avere un periodo di lucidità, e, successivamente, sviluppare
incontrollabile gonfiore cerebrale [McCrory e Berkovic 1998]. Trauma-innescata
ritardata edema cerebrale è stato associato con il CACNA1Amutazione missenso p.Ser218Leu [Kors et al 2001].
Nonostante i loro
fenotipi ben definite, SCA6, EA2, e FHM dimostrano sovrapposizione clinica:
Gli individui con SCA6 possono
presentarsi con atassia episodica. In uno studio, fino al 33% delle
persone fisiche con 21 o più CAG ripetizioni in CACNA1A aveva
caratteristiche episodiche abbastanza importanti da giustificare la
diagnosi di EA2 [Geschwind et al 1997].
In una famiglia con un CAG
repeat espansione, alcuni membri avevano atassia episodica e altri avevano
atassia progressiva; in tutti i colpiti membri anormale allele aveva 23 CAG ripetizioni [Jodice et al 1997].
In una famiglia con EA2, colpiti i membri aveva anche
emiplegia, e un membro affetto avuto emicrania durante gli episodi di
atassia [Jen 1999].
Atassia spinocerebellare
di tipo 6 (SCA6) è caratterizzata da insorgenza nell'età adulta, lentamente
progressiva atassia cerebellare, disartria e nistagmo. Il range di età di esordio
è da 19 a 71 anni. L'età media di insorgenza è tra i 43 ei 52
anni. L'età di esordio e quadro clinico varia anche all'interno della
stessa famiglia; fratelli e sorelle con la stessa dimensione full penetranzaallele possono differire
in età di insorgenza di ben 12 anni, o mostra, almeno inizialmente, un corso
episodico [Gomez et al 1997,Jodice et al 1997].
I sintomi iniziali sono
instabilità dell'andatura, inciampando, e lo squilibrio in circa il 90% degli
individui; il resto presente con disartria. I sintomi progredire
lentamente, e, infine, tutte le persone che hanno andatura atassia, degli arti
superiori mancanza di coordinazione, tremore intenzionale, e disartria. La
disfagia e soffocamento sono comuni.
Diplopia si verifica in
circa il 50% degli individui. Altri sperimentano disturbi visivi legati
alla difficoltà fissarsi su oggetti in movimento, così come nistagmo
orizzontale sguardo-evocato (70% -100%) e nistagmo verticale (65% -83%), che si
osserva in meno del 10% di quelli con altri forme di SCA [Yabe et al 2003]. Altre anomalie dei
movimenti oculari, tra cui periodici nistagmo alternante e nistagmo rimbalzo, sono
stati descritti [Hashimoto et al
2003].
Iperreflessia e risposte
plantari estensori si verificano in circa il 40% -50% delle persone con SCA6.
Segni dei gangli della
base, come la distonia e blefarospasmo, sono noti in fino al 25% degli
individui.
Mentazione viene
generalmente conservato. Formale test neuropsicologici in una serie non
hanno rivelato deficit cognitivi significativi [Globas et al 2003].
Gli individui con SCA6
non hanno reclami sensoriali, delle gambe senza riposo, rigidità, emicrania,
disturbi visivi primari, o atrofia muscolare.
L'aspettativa di vita
non è accorciato.
Gravidanza. La gravità della
malattia aumenta durante la gravidanza. Non sono stati riportati effetti
sulla vitalità del feto.
Neuropatologia. Neuropathologic
studi in individui con SCA6 hanno dimostrato sia selettiva degenerazione delle
cellule di Purkinje o una degenerazione combinata delle cellule di Purkinje e
granuli [Gomez et al 1997,Sasaki et al 1998].
Genotipo-fenotipo
Correlazioni
Individui eterozigoti. Anche se l'età di
insorgenza dei sintomi di SCA6 correla inversamente con la lunghezza della
ripetizione CAG espansa, la stessa ampia gamma di insorgenza è stato notato per
gli individui con 22 ripetizioni CAG, la malattia associata più comune allele[Gomez et al 1997,Schols et al 1998]. Nei pochi
individui con (CAG) 30 o (CAG) 33, l'inizio
è stato più tardi che in individui con (CAG) e 22 (CAG) 23[Matsuyama et al 1997,Yabe et al 1998].Un recente studio
retrospettivo ha mostrato anche una correlazione più stretta di età di esordio
con la somma delle due grandezze allele [Takahashi et al 2004].
La penetranza è quasi
del 100%, anche se i sintomi non possono comparire fino al settimo decennio.
Anticipazione
Espansioni di CACNA1A non
sono comunemente osservate in trasmissione da madre a
figlio; quindi, anticipazione non è stata
osservata in SCA6. L'età di esordio, la gravità, sintomi specifici, e la
progressione della malattia sono variabili e non può essere previsto
dalla storia familiare o CAG repeat
dimensioni.
Nomenclatura
Forme ereditarie di
atassia, una volta conosciuta come tipo di Holmes cerebellare degenerazione
corticale, e poi comeautosomica dominante tipo atassia
cerebellare III (atassia cerebellare puro), possono avere incluso SCA6.
Prevalenza
La prevalenza di SCA6
sembra variare per area geografica, presumibilmente in relazione agli effetti
fondatore. Stimato come la frazione di tutte le parentele con autosomica dominante atassia
spinocerebellare, tariffe per SCA6 sono 1% -2% in Spagna e in Francia, il 3% in
Cina, il 12% negli Stati Uniti, il 13% in Germania, e il 31% in Giappone.
La frequenza di
espansioni CACNA1A tra gli individui con atassia e senza
conoscere la storia familiare di atassia è stato
determinato pari al 5% in uno studio [Schols et al 1998] e il 43% in un
altro [Geschwind et al
1997]; tuttavia,
la morte prematura dei genitori può aver ostacolato l'accertamento completo di
tutti i casi (vedi atassia Panoramica).
Gli individui con
atassia spinocerebellare di tipo 6 (SCA6) possono presentare atassia
inspiegabile che fa parte della diagnosi differenziale maggiore di atassie
ereditarie ed acquisite (vedi atassia Panoramica).
Per stabilire l'entità
della malattia e la corretta gestione di un individuo con diagnosi di atassia
spinocerebellare di tipo 6 (SCA6), si consigliano le seguenti valutazioni:
Medico, neurologico, la
famiglia e la storia sociale
Esame neurologico, compreso
l'uso di una scala di valutazione da utilizzare annualmente per valutare
la progressione
MRI del cervello per misurare il
grado di atrofia del cervelletto o altre strutture
Ingoiare la valutazione del
rischio per l'aspirazione e la consulenza
Valutazione PT per valutare il
rischio di caduta, per determinare se la deambulazione assistita è
necessario, e per consigliare in materia di esercizio.
Consultazione Genetica medica
Trattamento delle
manifestazioni
La gestione è di
supporto.
Supplementi di vitamina sono
raccomandati, in particolare se l'apporto calorico è ridotto.
Acetazolamide può eliminare
episodi di atassia, ma non ritardare o rallentare la progressione
complessiva.
Soppressori vestibolari possono
ridurre vertigini e / o osscilopsia.
Consultazione Oftalmologia è
indicato per la gestione di rifrazione o chirurgica di diplopia.
Sebbene né l'esercizio, né la
terapia fisica steli la progressione di incoordinazione o debolezza
muscolare, colpitigli individui devono mantenere
l'attività. La terapia fisica dovrebbe essere mirata a massimizzare
il risarcimento e la forza.
Canne, bastoni da passeggio, e
camminatori aiutare a prevenire caduta. Modifica della casa con tali
convenienze come maniglie, sedili WC sollevato, e rampe per accogliere
sedie a motore può essere necessario.
Disturbi del linguaggio di
tanto in tanto si verificano e possono essere gestiti come in altri
contesti. Dispositivi di logopedia e di comunicazione quali
cuscinetti di scrittura e dispositivi basati su computer possono
beneficiare le persone con disartria.
Calibrati posate e ganci
spogliatoio aiutano a mantenere un senso di indipendenza.
Il controllo del peso è
importante perché l'obesità può esacerbare le difficoltà con la
deambulazione e mobilità.
Prima di disfagia diventa
fastidioso, esophagrams video possono identificare i comportamenti più
sicuri e la consistenza del cibo meno probabilità di innescare
aspirazione.
Clonopin può essere utilizzato
per disturbi del comportamento del sonno REM (RBD) a meno effetti sedativi
aumentano squilibrio nella mattina. Nota: RBD è rara nei pazienti con
SCA6.
Pressione positiva continua può
essere utilizzato per l'apnea del sonno.
Sorveglianza
Gli individui affetti
dovrebbero essere seguiti annualmente o semestralmente da un neurologo, con le
consultazioni in base alle esigenze fisiatra e fisica e / o terapista
occupazionale. Capacità di guida deve essere valutata da professionisti
periodicamente.
Agenti / Circostanze
da evitare
Agenti con sedativi /
proprietà ipnotiche come farmaci etanolo o alcuni possono produrre un aumento
della incoordinazione segnato.
Anche se la malattia si
manifesta raramente durante gli anni di fertilità, le misure per sostenere lo
squilibrio dovrebbe venire rafforzata in donne in gravidanza sintomatiche.
Terapie Sotto
inchiesta
Gazulla & Tintore [2007] hanno suggerito
gabapentin e pregabalin come potenziali agenti terapeutici.
Cerca ClinicalTrials.gov per l'accesso alle
informazioni su studi clinici per una vasta gamma di malattie e condizioni.
Altro
Farmaci-tremore che
controlla di solito non sono efficaci nel ridurre i tremori cerebellari.
I pazienti e le loro
famiglie dovrebbero essere informati sulla storia naturale, trattamento, modalità di
trasmissione, rischi genetici per altri membri della famiglia, e le
risorse consumer-oriented.
La consulenza genetica è
il processo di fornitura di individui e famiglie con informazioni sulla natura,
l'ereditarietà, e le implicazioni di malattie genetiche per aiutarli a prendere
decisioni personali e mediche informate.I seguenti sezione tratta la
valutazione del rischio genetico e l'uso della storia familiare e test genetici
per chiarire lo status genetico per i familiari.Questa sezione non
è destinata a affrontare tutte le questioni personali, culturali, etiche o che
gli individui possono affrontare o per sostituire la consultazione con una
genetica professionale. -ED.
Modalità di eredità
Atassia spinocerebellare
di tipo 6 (SCA6) è ereditata come autosomica dominante maniera.
Perché penetranza è al 100%, la maggior
parte degli individui con diagnosi di SCA6 hanno un affetto genitore.
Un probando con SCA6 può avere la
malattia come il risultato di un novo degenemutazione. La proporzione di casi
causata da mutazioni de novo del gene è sconosciuto.
Nota: La storia della famiglia può sembrare
negativo a causa del mancato riconoscimento del disturbo di membri della
famiglia, la morte precoce del genitore prima della comparsa dei sintomi, o
tardiva insorgenza della malattia nelinteressata genitore.
Il rischio per i fratelli e
sorelle di un affetto individuo dipende dallo
stato genetico dei probando genitori 's.
Se un genitore ha un CACNA1A espansa allele o un'altra mutazione, il rischio per ogni sib
di ereditare ilCACNA1A allele che causano malattie e
sviluppare la malattia è del 50%.
Se nessuno dei genitori
biologici del probando ha un CACNA1A causano
malattie allele rilevabile in DNA, si presume che i probandi
ha un de novogenemutazioni e il rischio per i
fratelli del probando dipende dalla probabilità di mosaicismo
germinale. Sebbene
non siano stati riportati casi di mosaicismo germinale, rimane una
possibilità.
Figli di un probando. Prole di affetti individui hanno una
probabilità del 50% di ereditare la CACNA1A alterato allele,e sviluppare la
malattia.
Cambiamenti in repeat
dimensione. Anche se cambia la dimensione di ripetizione possono
verificarsi in alleli SCA6 nelle generazioni successive, che sono molto più
rari rispetto a quelli di altri disturbi di espansione ripetizione.
Altri membri della
famiglia di un probando. Il rischio per gli
altri membri della famiglia dipende dallo status genetico dei genitori del
probando. Se un genitore ha il CACNA1A causano
malattie allele, i suoi o suoi
familiari sono a rischio.
Genetica Related Issues
Counseling
Prove di a rischio
adulti asintomatici. Prove di adulti a rischio di SCA6 è possibile utilizzando le
tecniche descritte nella ricerca di genetica
molecolare. Tale test non è utile nel predire l'età di esordio, la
gravità, il tipo di sintomi, o tasso di progressione in individui
asintomatici. Durante il test i soggetti a rischio per SCA6, un colpiti membro della
famiglia deve essere testato prima di confermare la diagnosi di SCA6. Test
per la mutazione responsabile
della malattia in assenza di sintomi o segni della malattia definite
è test predittivi. A rischio i membri
asintomatici familiari adulti possono cercare le prove, al fine di prendere
decisioni personali che lo riguardano riproduzione, questioni finanziarie, e
pianificazione della carriera. Altri possono avere diverse motivazioni tra
cui semplicemente "la necessità di sapere."Prove di asintomatici
membri adulti della famiglia a rischio di solito comporta interviste pre-test,
in cui i motivi per la richiesta del test, conoscenza dell'individuo della
SCA6, il possibile impatto dei risultati dei test positivi e negativi, e lo
stato neurologico sono valutati. Coloro che cercano di test dovrebbero
essere informati sui possibili problemi che possono incontrare per quanto
riguarda la salute, la vita, e la copertura assicurativa di invalidità,
l'occupazione e la discriminazione nei, e cambiamenti di interazione sociale e
familiare. Altri aspetti da considerare sono le implicazioni per lo stato
a rischio di altri membri della famiglia. Il consenso informato deve
essere procurato e le registrazioni mantenute riservate. Gli individui con
un risultato positivo hanno bisogno modalità di lungo periodo di follow-up e
valutazioni.
Prove di soggetti a
rischio asintomatici durante l'infanzia. Consensus sostiene che gli individui
di età inferiore ai 18 anni che sono a rischio per i disturbi ad insorgenza
nell'età adulta non dovrebbe avere test in assenza di sintomi. Gli
argomenti principali contro i test individui asintomatici prima dei 18 anni
sono che rimuove la loro scelta di sapere o non sapere queste informazioni, si
prospetta la possibilità di stigmatizzazione all'interno della famiglia e in
altri contesti sociali, e potrebbe avere gravi implicazioni educative e di
carriera. Gli individui di età inferiore ai 18 anni che sono sintomatici
di solito sono interessati ad avere una diagnosi specifica stabilita. Per
ulteriori informazioni, consultare anche la Società Nazionale di Genetic
Consulenti presa di posizione sui test genetici di
minori per le condizioni ad insorgenza nell'età adulta e la American Academy of
Pediatrics e dell'American College of Medical Genetics e Genomicadichiarazione
politica: questioni
etiche e politiche nei test genetici e lo screening dei bambini.
Considerazioni in
famiglie con un apparente de novomutazione. Quando nessuno dei
genitori di un probando con una autosomica dominante condizione ha
la mutazione responsabile
della malattia o la prova della malattia, è possibile che i probandi ha una
mutazione de novo. Tuttavia, possibili spiegazioni non medici,
tra cui la paternità
alternativo o di maternità (ad esempio, con la riproduzione assistita) o
l'adozione riservate potrebbero anche essere esplorate.
Pianificazione
famigliare
Il momento ottimale per la
determinazione del rischio genetico e discussione della disponibilità di
test prenatale è prima della gravidanza.
È opportuno offrire una consulenza
genetica (compresa
la discussione dei rischi potenziali per la prole e le opzioni di
riproduzione) di giovani adulti che sono affetti oa rischio.
Banking DNA è la conservazione
di DNA (tipicamente estratto da globuli bianchi) per un possibile uso
futuro. Poiché è probabile che la metodologia di prova e la nostra
comprensione dei geni, mutazioni, e le malattie migliorerà in futuro, occorre
tenere in considerazione per bancaria DNA di affetti individui.
Test prenatale
Se la mutazione responsabile
della malattia è stato identificato in un colpiti membro della
famiglia, test prenatale per gravidanze a rischio è possibile mediante
l'analisi di DNA estratto da
cellule fetali ottenuti mediante l'amniocentesi (solitamente eseguita presso
gestazione ~ 15-18 settimane) o dei villi coriali campionamento (di solito
eseguita presso gestazione ~ 10-12 settimane).
Nota: l'età gestazionale
è espresso in settimane mestruali calcolate sia a partire dal primo giorno
dell'ultimo periodo mestruale normale o da misure ad ultrasuoni.
Le richieste di diagnosi prenatale di malattie (in
genere) ad insorgenza nell'età adulta non sono comuni. Possono esistere
differenze di prospettiva tra i professionisti medici e all'interno delle
famiglie per quanto riguarda l'uso di test prenatali, in particolare se il test
viene presa in considerazione ai fini della interruzione della gravidanza o di
diagnosi precoce.Sebbene la maggior parte centri prenderebbero in
considerazione decisioni su test prenatale per essere la scelta dei genitori,
la discussione di questi problemi è appropriato.
Diagnosi genetica
preimpianto (PGD) può essere un'opzione per alcune famiglie in cui la malattia che causa la
mutazione è
stato identificato / espansione.
Personale GeneReviews ha
selezionato le seguenti organizzazioni malattia-specifici e / o di supporto
ombrello e / o registri per il beneficio di soggetti con questo disturbo e le
loro famiglie.GeneReviews non è responsabile per le informazioni
fornite da altre organizzazioni.Per informazioni sui criteri di
selezione, clicca qui.
E-mail:Questo indirizzo email è protetto dagli spambots. È necessario abilitare JavaScript per vederlo.; Questo indirizzo email è protetto dagli spambots. È necessario abilitare JavaScript per vederlo.
E-mail:Questo indirizzo email è protetto dagli spambots. È necessario abilitare JavaScript per vederlo.; Questo indirizzo email è protetto dagli spambots. È necessario abilitare JavaScript per vederlo.; Questo indirizzo email è protetto dagli spambots. È necessario abilitare JavaScript per vederlo.
Le informazioni
contenute in tabelle Genetica Molecolare e OMIM può differire da quello in
altre parti del GeneReview: tavoli può contenere informazioni più recenti.- ED.
I
dati sono compilati dai seguenti riferimenti standard: simbolo del gene
da HGNC; cromosomico locus,
nome luogo, regione critica, complementazione gruppo da OMIM; nome
proteine UniProt. Per una
descrizione di database (Locus specifico, HGMD) a cui vengono forniti i link,
clicca qui.
CALCIO CHANNEL,
voltaggio-dipendente, P / Q TIPO, ALPHA-1A SUBUNITA; CACNA1A
Gene struttura.CACNA1A composto
da 47 esoni. Per un riepilogo dettagliato delle gene informazioni e
proteine, vedere tabella A, Gene Simbolo.
Varianti alleliche
normali. Una
ripetizione CAG polimorfici in all'estremità 3 'del gene avviene
all'interno di una porzione del gene precedentemente pensato per essere solo
non codificante. L'identificazione di espansioni di questo CAG repeat
associata con autosomica dominante atassia è stata
accompagnata dal riconoscimento di una nuova forma lunga giuntura della alfa 1A
mRNA in cui il quadro di lettura include CAG repeat
tradotti in residui di glutammina. I CAG ripetizioni vanno da (CAG)
a 4 (CAG) 18.
Sono state
riportate le varianti alleliche patogeni. Associata a malattia
alleli CAG-repeat che vanno da (CAG) 21(CAG) 33. Il
più comune allele è (CAG) 22. Un
individuo con un (CAG) 20 allele ha atassia
episodica [Jodice et al 1997].
Normale prodotto del gene.CACNA1A codifica
per due proteine distinte, una subunità tipo a1A che serve come la subunità poro formazione di un canale del calcio P / Q-tipo
voltaggio-dipendente (recensione in Greenberg [1997]), e unfattore di
trascrizione, α1ACT, che trasloca nel nucleo e atti a migliorare
l'espressione di diversi geni espressi neuronally[Du et al 2.013]. Sia Alpha-1A
(alcuni di splicing-forme) e α1ACT portano CAG repeat polimorfici che si
espande in SCA6.
Canali del calcio
voltaggio-dipendenti sono costituiti da subunità beta e gamma di
accessori-δ. subunità tipo a1A sono glicoproteine di membrana di circa
2400 amminoacidi di lunghezza in cui struttura primaria predice la presenza di
quattro domini omologhi, ciascuno costituito da sei domini transmembrana e un
anello P formano pori. Canali del calcio P / Q-type sono tensione attivati
high-canali del calcio
presenti soprattutto sui neuroni e hanno espresso ad alti livelli nelle cellule
dei granuli e cellule di Purkinje della corteccia cerebellare. Il loro
ruolo principale si crede di essere nella trasmissione sinaptica. Il
α1 (2.1), precedentemente tipo a1A, subunità è il principale formano pori
subunità del (tipo PQ) CaV 2.1 canali del calcio
voltaggio-dipendenti. CACNA1A dà luogo a diversi mRNA splicing
alternativo di circa 7-8 kb [Ophoff et al 1996 ]. I
polipeptidi previste vanno da 195 a 270 kd e variano in sequenza all'interno e
nel terminale carbossi. La scoperta del CAG repeat polimorfici in
all'estremità 3 'del gene è stata associata
con l'individuazione di una nuova forma lunga giuntura del tipo a1A mRNA [Zhuchenko et al 1997]. Nella forma splice
lunga, inclusione di nucleotidi complementari alla fine dell'esone 46 elimina uno
stop codone e pone ulteriori
237 nucleotidi 3 'sequenza, compresa la ripetizione CAG polimorfici, nel telaio
traslazionale. La ripetizione CAG codifica un tratto di residui di
glutammina, in cui tipo alleli selvatici vanno da quattro a 18 glutammato di
lunghezza. La funzione delle diverse forme splice dei prodotti
genici CACNA1A resta da dimostrare, anche se le differenze
sono state misurate in siti accettori di fosforilazione.
α1ACT è codificato
come una proteina separato all'interno del 'porzione 3 della forma lungo
giunzione di tipo a1A subunità mRNA. The α1ACT
polypeptide is translated from the α1A mRNA as a separate protein under the control of a cellular
internal ribosomal entry site (IRES). α1ACT is a transcriptional protein that is translocated to nuclei
and binds and enhances expression via a conserved AT-rich motif on several
genes expressed in Purkinje cells. α1ACT expressed without α1A subunits accelerates neurite outgrowth in cultured neuronal
cells and normalizes Purkinje cell dendrites and innervation when expressed in α1A knockout
mice. α1ACT
polypeptide bears the polymorphic polyglutamine tract [Du et al 2013 ].
Abnormal gene
product . The expanded CAG repeat in CACNA1A in SCA6
codes for an expanded polyglutamine tract present in both the carboxy terminus
of a long splice form of the α1A subunit of the P/Q-type calcium channel [Zhuchenko et al 1997 ] and
the α1ACT protein.
While there is no consistent effect of the expanded polyglutamine tract on P/Q
channel function, the polyqlutamine expansion alters gene binding,
impairs transcription
factor function and is toxic to cells expressing the α1ACT.
Committee
on Bioethics, Committee on Genetics, and American College of Medical
Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and
policy issues in genetic testing and screening of children. Available online . 2013. Accessed 2-18-15.
[ PubMed ]
National
Society of Genetic Counselors. Position statement on genetic testing of
minors for adult-onset disorders. Available online . 2012. Accessed
11-17-14.
Literature Cited
Alonso
I, Barros J, Tuna A, Coelho J, Sequeiros J, Silveira I, Coutinho P.
Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic
migraine caused by a unique CACNA1A missense mutation in patients from a
large family. Arch
Neurol. 2003; 60 :610–4. [ PubMed ]
Baloh
RW, Yue Q, Furman JM, Nelson SF. Familial episodic ataxia: clinical
heterogeneity in four families linked to chromosome 19p. Ann Neurol. 1997; 41 :8–16. [ PubMed ]
Battistini
S, Stenirri S, Piatti M, Gelfi C, Righetti PG, Rocchi R, Giannini F,
Battistini N, Guazzi GC, Ferrari M, Carrera P. A new CACNA1A gene mutation
in acetazolamide-responsive familial hemiplegic migraine and
ataxia. Neurology. 1999; 53 :38–43. [ PubMed ]
Chen
H, Piedras-Renteria ES. Altered frequency-dependent inactivation and
steady-state inactivation of polyglutamine-expanded alpha1A in SCA6. Am J Physiol Cell
Physiol. 2007; 292 :C1078–86. [ PubMed ]
Craig
K, Keers SM, Archibald K, Curtis A, Chinnery PF. Molecular epidemiology of
spinocerebellar ataxia type 6. Ann
Neurol. 2004; 55 :752–5. [ PubMed ]
Cricchi
F, Di Lorenzo C, Grieco GS, Rengo C, Cardinale A, Racaniello M, Santorelli
FM, Nappi G, Pierelli F, Casali C. Early-onset progressive ataxia
associated with the first CACNA1A mutation identified within the I-II
loop. J Neurol Sci. 2007; 254 :69–71. [ PubMed ]
Du XL,
Wang J, Zhu H, Rinaldo L, Lamar K, Palmenberg AC, Hansel C, Gomez CM. Second cistron inCACNA1A gene
encodes a transcription factor mediating cerebellar development and
SCA6. Cell. 2013; 154:118–33. [ PMC free
article ]
[ PubMed ]
Ducros
A, Denier C, Joutel A, Cecillon M, Lescoat C, Vahedi K, Darcel F, Vicaut
E, Bousser MG, Tournier-Lasserve E. The clinical spectrum of familial
hemiplegic migraine associated with mutations in a neuronal calcium
channel. N
Engl J Med. 2001; 345 :17–24. [ PubMed ]
Ducros
A, Denier C, Joutel A, Vahedi K, Michel A, Darcel F, Madigand M, Guerouaou
D, Tison F, Julien J, Hirsch E, Chedru F, Bisgard C, Lucotte G, Despres P,
Billard C, Barthez MA, Ponsot G, Bousser MG, Tournier-Lasserve E.
Recurrence of the T666M calcium channel CACNA1A gene mutation in familial
hemiplegic migraine with progressive cerebellar ataxia. Am J Hum
Genet. 1999; 64 :89–98. [ PMC free
article ]
[ PubMed ]
Friend
KL, Crimmins D, Phan TG, Sue CM, Colley A, Fung VS, Morris JG, Sutherland
GR, Richards RI. Detection of a novel missense mutation and second
recurrent mutation in the CACNA1A gene in individuals with EA-2 and
FHM. Hum
Genet. 1999; 105 :261–5. [ PubMed ]
Gazulla
J, Tintore MA. The P/Q-type voltage-dependent calcium channel as
pharmacological target in spinocerebellar ataxia type 6: gabapentin and
pregabalin may be of therapeutic benefit. Med
Hypotheses. 2007;68 :131–6. [ PubMed ]
Geschwind
DH, Perlman S, Figueroa KP, Karrim J, Baloh RW, Pulst SM. Spinocerebellar
ataxia type 6. Frequency of the mutation and genotype-phenotype
correlations. Neurology. 1997; 49 :1247–51. [ PubMed ]
Globas
C, Bosch S, Zuhlke Ch, Daum I, Dichgans J, Burk K. The cerebellum and
cognition. Intellectual function in spinocerebellar ataxia type 6
(SCA6). J Neurol. 2003; 250 :1482–7. [ PubMed ]
Gomez
CM, Thompson RM, Gammack JT, Perlman SL, Dobyns WB, Truwit CL, Zee DS,
Clark HB, Anderson JH. Spinocerebellar ataxia type 6: gaze-evoked and
vertical nystagmus, Purkinje cell degeneration, and variable age of
onset. Ann
Neurol. 1997; 42 :933–50. [ PubMed ]
Greenberg
DA. Calcium channels in neurological disease. Ann
Neurol. 1997; 42 :275–82. [ PubMed ]
Hashimoto
T, Sasaki O, Yoshida K, Takei Y, Ikeda S. Periodic alternating nystagmus
and rebound nystagmus in spinocerebellar ataxia type 6. Mov
Disord. 2003; 18 :1201–4. [ PubMed ]
Ikeuchi
T, Takano H, Koide R, Horikawa Y, Honma Y, Onishi Y, Igarashi S, Tanaka H,
Nakao N, Sahashi K, Tsukagoshi H, Inoue K, Takahashi H, Tsuji S.
Spinocerebellar ataxia type 6: CAG repeat expansion in alpha1A voltage-
dependent calcium channel gene and clinical variations in Japanese
population. Ann Neurol. 1997; 42:879–84. [ PubMed ]
Ishikawa
K, Tanaka H, Saito M, Ohkoshi N, Fujita T, Yoshizawa K, Ikeuchi T,
Watanabe M, Hayashi A, Takiyama Y, Nishizawa M, Nakano I, Matsubayashi K,
Miwa M, Shoji S, Kanazawa I, Tsuji S, Mizusawa H. Japanese families with
autosomal dominant pure cerebellar ataxia map to chromosome 19p13.1-p13.2
and are strongly associated with mild CAG expansions in the
spinocerebellar ataxia type 6 gene in chromosome 19p13.1.Am J Hum
Genet. 1997; 61 :336–46. [ PMC free
article ]
[ PubMed ]
Jen
J. Calcium channelopathies in the central nervous system. Curr Opin
Neurobiol. 1999; 9 :274–80. [ PubMed ]
Jiang
H, Tang B, Xia K, Zhou Y, Xu B, Zhao G, Li H, Shen L, Pan Q, Cai F.
Spinocerebellar ataxia type 6 in Mainland China: molecular and clinical
features in four families. J
Neurol Sci. 2005; 236 :25–9. [ PubMed ]
Jodice
C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L,
Francia A, Spadaro M, Pierelli F, Salvi F, Ophoff RA, Frants RR, Frontali
M. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6)
due to CAG repeat expansion in the CACNA1A gene on chromosome
19p. Hum Mol Genet. 1997; 6:1973–8. [ PubMed ]
Katayama
T, Ogura Y, Aizawa H, Kuroda H, Suzuki Y, Kuroda K, Kikuchi K. Nineteen
CAG repeats of the SCA6 gene in a Japanese patient presenting with
ataxia. J
Neurol. 2000; 247 :711–2. [ PubMed ]
Kordasiewicz
HB, Gomez CM. Molecular pathogenesis of spinocerebellar ataxia type
6. Neurotherapeutics.2007; 4 :285–94. [ PubMed ]
Kors
EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, Love
S, van den Maagdenberg AM, Haan J, Frants RR, Ferrari MD. Delayed cerebral
edema and fatal coma after minor head trauma: role of the CACNA1A calcium
channel subunit gene and relationship with familial hemiplegic
migraine. Ann
Neurol. 2001;49 :753–60. [ PubMed ]
Mariotti
C, Gellera C, Grisoli M, Mineri R, Castucci A, Di Donato S. Pathogenic
effect of an intermediate-size SCA-6 allele (CAG)(19) in a homozygous
patient. Neurology. 2001; 57 :1502–4. [ PubMed ]
Matsumura
R, Futamura N, Fujimoto Y, Yanagimoto S, Horikawa H, Suzumura A,
Takayanagi T. Spinocerebellar ataxia type 6. Molecular and clinical features
of 35 Japanese patients including one homozygous for the CAG repeat
expansion. Neurology. 1997; 49 :1238–43. [ PubMed ]
Matsuyama
Z, Kawakami H, Maruyama H, Izumi Y, Komure O, Udaka F, Kameyama M, Nishio
T, Kuroda Y, Nishimura M, Nakamura S. Molecular features of the CAG
repeats of spinocerebellar ataxia 6 (SCA6). Hum Mol
Genet. 1997; 6 :1283–7. [ PubMed ]
McCrory
PR, Berkovic SF. Second impact syndrome. Neurology. 1998; 50 :677–83. [ PubMed ]
Montagna
P. Molecular genetics of migraine headaches: a review. Cephalalgia. 2000; 20 :3–14. [ PubMed ]
Ophoff
RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin
JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen
GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine
and episodic ataxia type-2 are caused by mutations in the Ca2+ channel
gene CACNL1A4.Cell. 1996; 87 :543–52. [ PubMed ]
Pujana
MA, Corral J, Gratacos M, Combarros O, Berciano J, Genis D, Banchs I,
Estivill X, Volpini V. Spinocerebellar ataxias in Spanish patients:
genetic analysis of familial and sporadic cases. The Ataxia Study
Group. Hum Genet. 1999; 104 :516–22. [ PubMed ]
Riess
O, Schols L, Bottger H, Nolte D, Vieira-Saecker AM, Schimming C, Kreuz F,
Macek M, Krebsova A, Macek M, SenKlockgether T, Zuhlke C, Laccone FA. SCA6 is caused by
moderate CAG expansion in the alpha1A-voltage- dependent calcium channel
gene. Hum Mol
Genet. 1997; 6 :1289–93. [ PubMed ]
Sasaki
H, Kojima H, Yabe I, Tashiro K, Hamada T, Sawa H, Hiraga H, Nagashima K.
Neuropathological and molecular studies of spinocerebellar ataxia type 6
(SCA6). Acta
Neuropathol (Berl) 1998; 95 :199–204. [PubMed ]
Schols
L, Kruger R, Amoiridis G, Przuntek H, Epplen JT, Riess O. Spinocerebellar
ataxia type 6: genotype and phenotype in German kindreds. J Neurol Neurosurg
Psychiatry. 1998; 64 :67–73. [ PMC free
article ]
[ PubMed ]
Shimazaki
H, Takiyama Y, Sakoe K, Amaike M, Nagaki H, Namekawa M, Sasaki H, Nakano
I, Nishizawa M. Meiotic instability of the CAG repeats in the SCA6/CACNA1A
gene in two Japanese SCA6 families. J Neurol
Sci. 2001; 185 :101–7. [ PubMed ]
Shizuka
M, Watanabe M, Ikeda Y, Mizushima K, Okamoto K, Shoji M. Molecular
analysis of a de novo mutation for spinocerebellar ataxia type 6 and
(CAG)n repeat units in normal elder controls. J Neurol
Sci. 1998;161 :85–7. [ PubMed ]
Stevanin
G, Durr A, David G, Didierjean O, Cancel G, Rivaud S, Tourbah A, Warter
JM, Agid Y, Brice A. Clinical and molecular features of spinocerebellar
ataxia type 6. Neurology. 1997; 49 :1243–6. [ PubMed ]
Takahashi
H, Ishikawa K, Tsutsumi T, Fujigasaki H, Kawata A, Okiyama R, Fujita T,
Yoshizawa K, Yamaguchi S, Tomiyasu H, Yoshii F, Mitani K,
Shimizu N, Yamazaki M, Miyamoto T, Orimo T, Shoji S, Kitamura K, Mizusawa
H. A clinical and genetic study in a large cohort of patients with
spinocerebellar ataxia type 6. J Hum
Genet. 2004; 49 :256–64. [ PubMed ]
Tonelli
A, D'Angelo MG, Salati R, Villa L, Germinasi C, Frattini T, Meola G,
Turconi AC, Bresolin N, Bassi MT. Early
onset, non-fluctuating spinocerebellar ataxia and a novel missense
mutation in CACNA1A gene. J Neurol
Sci. 2006; 241 :13–7. [ PubMed ]
Yabe
I, Sasaki H, Matsuura T, Takada A, Wakisaka A, Suzuki Y, Fukazawa T,
Hamada T, Oda T, Ohnishi A, Tashiro K. SCA6 mutation analysis in a large
cohort of the Japanese patients with late-onset pure cerebellar
ataxia. J Neurol
Sci. 1998; 156 :89–95. [ PubMed ]
Yabe
I, Sasaki H, Takeichi N, Takei A, Hamada T, Fukushima K, Tashiro K.
Positional vertigo and macroscopic downbeat positioning nystagmus in
spinocerebellar ataxia type 6 (SCA6). J
Neurol. 2003; 250 :440–3. [ PubMed]
Yue
Q, Jen JC, Nelson SF, Baloh RW. Progressive ataxia due to a missense
mutation in a calcium-channel gene.Am J Hum
Genet. 1997; 61 :1078–87. [ PMC free article ]
[ PubMed ]
Zhuchenko
O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB,
Subramony SH, Zoghbi HY, Lee CC. Autosomal
dominant cerebellar ataxia (SCA6) associated with small polyglutamine
expansions in the alpha 1A-voltage-dependent calcium channel. Nat
Genet. 1997; 15 :62–9. [ PubMed ]
Lettura consigliata
1.Zoghbi HY, Orr HT.
Spinocerebellar ataxias. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW,
Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online
Metabolic and Molecular Bases of Inherited Disease (OMMBID).2015. New York, NY: McGraw-Hill. Chap 226.
Lo scopo di questa pagina è
quello di considerare i risultati in particolare dell’ Infarto
del ponte. .
La
definizione di ictus bulbo pontino viene utilizzata per indicare quella
particolare patologia che si verifica proprio nel tratto che separa il bulbo
dal ponte, il cosiddetto solco bulbo pontino, e da cui arrivano le informazioni
inerenti al gusto, al tatto e all’udito. Generalmente l’ictus si verifica
quando le arterie che portano sangue al cervello vengono ostruite da un trombo
o da un embolo, e nel caso dell’ictus al bulbo pontino si verifica una mancanza
di informazioni sensoriali che può compromettere le funzioni di una parte del
corpo.
L’AICA
vascolarizza la porzione laterale del cervelletto anteroinferiore (incluso il
flocculo) e la porzione dorso laterale del ponte inferiore (compreso il
peduncolo cerebellare medio), Dall’AICA origina l’arteria labirintica la quale
si suddivide in due rami terminali: l’arteria vestibolare anteriore (che irrora
l’utricolo, il canale semicircolare anteriore ed orizzontale) e l’arteria
cocleare comune (che irrora coclea, sacculo ed il canale semicircolare
posteriore). Nel 15 % dei casi l’arteria labirintica origina direttamente
dall’arteria basilare.
Se è
coinvolta isolatamente l’arteria vestibolare anteriore, l’ischemia si può
presentare solo o prevalentemente con sintomi vestibolari periferici quali
vertigini, nausea, vomito ed instabilità. Se viene coinvolta l’arteria cocleare
o l’arteria labirintica ai precedenti sintomi vestibolari, si associano
ipoacusia ed acufeni. Una ischemia isolata del labirinto è pertanto
difficilmente differenziabile da altre patologie periferiche benigne, anche se
l’esordio acuto, l’età, la presenza di fattori di rischio vascolare e,
soprattutto, cli altri segni e sintomi neurologici, possono indirizzare verso
una patogenesi ischemica.
Infatti,
se l’ischemia coinvolge il settore pontino dorso-laterale (figura 6) possono
associarsi, omolateralmente alla lesione, paralisi periferica del faciale,
deficit di sindrome di Horner, dismetria e controlateralmente, perdita della
sensibilità termodolorifica brachio-crurale. I sintomi che la differenziano
dalla sindrome di Wallenberg sono la presenza di ipoacusia ed acufeni e
l’assenza di disfagia e disartria, abduzione dell’occhio o dei movimenti
coniugati orizzontali, ipoestesia al volto,
Come
riportato sopra, in caso di ischemia bulbare laterale (territorio PICA) o
pontina laterale (AICA), la sindrome neurologica è tipica e cli facile
identificazione clinica, ma, la presenza di sindromi parziali, può essere
ingannevole ed erroneamente interpretata come “labirintite’ È pertanto
consigliabile eseguire accertamenti di neuroimmoging qualora la sindrome
vertiginosa sia ad esordio acuto, prolungata e senza ipoacusia, soprattutto in
pazienti con fattori cli rischio cerebrovascolare, anche nei casi in cui
l’esame neurologico non evidenzi sintomi/segni associati.
L'occlusione
di AICA è considerata rara, ma generalmente si traduce in una sindrome pontino
laterale, nota anche come sindrome AICA. I sintomi includono: improvvisa
comparsa di vertigini e vomito, nistagmo, disartria, cadendo a lato della
lesione (a causa di danni ai nuclei vestibolari), e una varietà di
caratteristiche omolaterali compresi hemiataxia, la perdita di tutte le
modalità di sensazione del volto (dovuto per danni al preside sensoriale nuclei
del trigemino), paralisi facciale (a causa di danni al nucleo delviso), e la
perdita di udito e tinnito (a causa di danni ai nuclei cocleari). [2] Esiste
anche la perdita del dolore e della temperatura sensazione dal gli arti e del
tronco controlaterale, che può portare a confusione diagnostica con Sindrome di
Wallenberg, che dà anche luogo a "incrociate", segni neurologici, ma
normalmente non causano sintomi cocleari, grave paralisi facciale o perdita di
sensibilità facciale multimodale.
A
tal fine la RM encefalo (soprattutto con sequenze pesate in diffusione, utili
per evidenziare lesioni ischemiche recenti) è molto più sensibile della IC per
evidenziare ischemie di piccole dimensioni Nella stessa seduta la RM può essere
completata con lo studio dei vasi (angio RM) per documentare le anomalie steno
occlusive del sistema verebro-basilare. Una Volta determinata l’eziologia
ischemica della sintomatologia vertiginosa, è importante stabilire la
patogenesi dell’evento cerebrovascolare (aterotrombotico embolico, dissecazione
arteriosa). A riguardo, si segnala come l’ecodoppler non sia sufficientemente
informativo nello studio del sistema vertebro basilare, in quanto non consente
una ottimale valutazione del distretto intracranico ove si localizzano
prevalentemente le lesioni aterosclerotiche della vertebrale. Per escludere
fonti emboligene cardiache, sono utili ECC ecocardiografia transtoracica e
transesofagea.
Cause di ictus del tronco
encefalico - Le basi
l’ictus (Stroke) può
essere ischemico (mancanza di flusso sanguigno) o emorragico
(fuoriuscita di sangue nel cervello).
FATTORI DI RISCHIO PER TIA E L’ICTUS
To some extent, one can
predict risk of stroke.
In una certa misura, si può predire il rischio di ictus. Risk factors that are well known include: I fattori
di rischio che sono ben noti includono:
High Blood Pressure
(2)
Alta pressione sanguigna (2)
Collagen Vascular
disease
Malattie vascolari del collagene
Heart problem such as
atrial fibrillation (1.5) or old infarction (2.7) Problema
di cuore come la fibrillazione atriale (1.5) o vecchio infarto (2,7)
**Numbers in () are derived from
Whisnant et al, 1996** I
numeri in () sono derivati da Whisnant et al, 1996 Rischio di
pressione sanguigna elevata è ripida e chiaro.
Rischio dovuto
alla pressione sanguigna. Risk from elevated blood
pressure is steep and clear.Ad esempio, nello studio UK TIA, il rischio di recidiva di
ictus è aumentato del 28% per ogni aumento di 10 mm Hg della pressione
sistolica tra 130 e 160 (Farrell et al, 1991).
Although
LDL-cholesteral, HDL cholesteral seems to reduce risk of stroke (Sacco et al,
2001).Anche se
LDL-colesterolo, HDL colesterolo sembra ridurre il rischio di ictus (Sacco et
al, 2001).If your HDL cholesterol is > 35, then subtract one risk factor (a
negative risk factor).Se il
colesterolo HDL è> 35, bisogna sottrarre un fattore di rischio (un fattore
di rischio negativo).Mitral valve prolapse is not a significant risk factor, overall (0.8
risk).
Il prolasso
della valvola mitrale non è un fattore di rischio significativo, nel complesso
(rischio x 0,8).TIA is a very strong risk factor for stroke (5.6 x risk).TIA è un fattore di
rischio molto forte per l'ictus (rischio x 5.6).In general, relative risk
for most of the above factors decreases with age (Whisnant et al, 1996),
lending support for a unaggressive approach to risk factors in individuals of
advanced age.In
generale, il rischio relativo per la maggior parte dei fattori di cui sopra
diminuisce con l'età (Whisnant et al, 1996), sostenendo un approccio non
aggressivo per i fattori di rischio in individui di età avanzata.
Risk from
cholesterol can be further stratified into three groups, based on LDL (total
cholesterol - HDL)-(triglycerides).Rischio da colesterolo può essere ulteriormente
stratificata in tre gruppi, sulla base LDL (colesterolo totale - HDL) -
(trigliceridi).
LDLLDL
Risk
FactorsFattori di rischio
Risk
Level for vascular diseaseLivello di rischio per malattia vascolare
<
130
<130
None Nessuno
Low Basso
130-159 130-159
Less
than 2
Meno di 2
Moderate Moderato
>130 > 130
more
than 2
più di 2
High Alto
Controllable
risk factors include being overweight, having high (> 140/90) or low blood pressure,
heart disease, diabetes and smoking.Fattori di rischio controllabili comprendono il
sovrappeso, con elevata (> 140/90) o bassa pressione sanguigna, malattie
cardiache, diabete e fumo.Atrial fibrillation is a particularly
important risk factor -- stroke occurs in 4.5% of untreated patients with
atrial fibrillation per year.La fibrillazione atriale è un fattore di rischio
particolarmente importante – l’ictus si verifica nel 4,5% dei pazienti trattati
con fibrillazione atriale all'anno.While uncommon, chiropractic neck
manipulations can cause compression or tears of the vertebral arteries (Vibert
et al, 1993; Smith et al, 2003), and for this reason, maneuvers involving neck
"cracking" should be specifically avoided in individuals with
vertigo.Mentre ,
le manipolazioni chiropratiche del collo possono causare compressione o rotture
delle arterie vertebrali (Vibert et al, 1993; Smith et al, 2003) e per questo
motivo, le manovre "cracking" che coinvolgono il collo devono essere
specificamente evitati nei soggetti con vertigine.Whiplash
injuries can also damage the vertebral arteries as the arteries traverse the
vertebrae of the neck.
Il colpo
di frusta può anche danneggiare le arterie vertebrali che passano attraversano
le vertebre del collo.
L’ictus
ischemico è causato da ostruzione dei vasi sanguigni. L’ostruzione
può provenire da una sorgente distante - come ad esempio un coagulo dal cuore –
che poi viene chiamato "embolo". I blocchi possono derivare da
coagulazione dall'interno dei vasi - che poi sono chiamati "trombi".
Gli ictus trombotici sono più spesso attribuiti a accumulo di colesterolo
all'interno delle pareti del vaso sanguigno, che producono turbolenza e ispessimento
irregolare della parete, con successiva e conseguente formazione di coaguli.
Un'altra causa comune di ictus trombotico è una caduta di pressione sanguigna
- come potrebbe essere ad esempio causata da un arresto cardiaco di alcuni
minuti. Al di fuori di queste due categorie generali (emboli e trombi)ci possono
essere solo fonti occasionali di ictus ischemico Ad esempio, causati da
ostruzione meccanica dei vasi sanguigni – come potrebbe accadere durante una manipolazione
chiropratica del collo ad alta velocità o qualche altro evento che
causa un movimento collo molto forte. Si sono riscontrati (ad esempio), ictus
in pazienti subito dopo che gli stessi erano stati sulle montagne russe.
Ictus
emorragici - fuoriuscita di sangue all'interno del cervello - sono più
comunemente causate da pressione sanguigna troppo alta. Altre possibili
cause comprendono irregolarità nella coagulazione del sangue, danni alle pareti
dei vasi sanguigni da vari processi (compreso l'ictus ischemico). Nel
tronco encefalico, il circuito è stretto e gli ictus emorragici sono spesso
devastanti.
Dissection of vertebral artery (cutoff of left sided vessel).Dissezione
dell'arteria vertebrale .Immagine gentilmente concessa da Ruth Ramsey, MD Sul
lato destro è un'altra immagine di una dissezione vertebrale in un altro
paziente. The vertebral artery on the left side is
smaller and irregularly filled. L'arteria vertebrale sul lato sinistro
è più piccola e irregolare riempito.
Ictus
ischemici sono causati da ostruzione dei vasi sanguigni. I
blocchi possono provenire da una sorgente distante - come un coagulo dal cuore
- poi sono chiamati "emboli". I blocchi possono derivare da
coagulazione dall'interno - poi sono chiamati "trombi". Ictus
trombotici sono più spesso attribuiti a accumulo di colesterolo all'interno
delle pareti del vaso sanguigno, producendo una turbolenza e irruvidimento
della parete, sulla quale si forma un coagulo.Un'altra causa comune di ictus
trombotico è una caduta di pressione sanguigna mantenuto nel - come potrebbe
essere causa di un arresto cardiaco per alcuni minuti. Ci sono solo fonti
occasionali di ictus ischemico di fuori di queste due categorie generali
(emboli e trombi). Ad esempio, i tratti possono essere causati da
ostruzione meccanica dei vasi sanguigni - che potrebbe accadere durante una manipolazione
chiropratica del collo alta velocità , o qualche altro evento che
causa un movimento collo molto forte. Abbiamo incontrato i pazienti che
hanno subito ictus (come esempio), dopo montagne russe.
Ictus emorragici -
fuoriuscita di sangue all'interno del cervello - sono più comunemente causate
da troppo alta pressione sanguigna. Altre possibili cause comprendono
irregolarità nella coagulazione del sangue, danni alle pareti dei vasi
sanguigni da vari processi (compreso l'ictus ischemico). Nel tronco
encefalico, il circuito è stretto e ictus emorragico sono spesso devastanti.
Infarto del ponte -Emorragia
Pontine.
Questo
è un evento catastrofico, Un’occlusione dell’AICA causa un infarto della
regione pontino-bulbare dorso-laterale e cerebellare infero-laterale (cfr Figg.
20 e 21). tipicamente in iperteso. Nell’85°/o dei casi l’ischemia
interessa anche l’arteria uditiva interna. per cui la sintomatologia è
costituita da una sindrome vestibolare deficitaria periferica con ipoacusia
improvvisa, concomitante ai segni dell’interessamento cerebellare o dei nuclei
dei nervi cranici eventualmente coinvolti dall’ischemia (per es. VII). Si
presenta con coma, tetraplegia, piccole pupille reattive e assenti movimenti
oculari orizzontali. Nella maggior parte dei pazienti tetraplegici un
ematoma nel mezzo del ponte è centrato al bivio del tegmento e la base
Pontis. Una caratteristica costante è il bobbing oculare. Emorragie
tegmentale laterali presentano con 1 1/2 sindrome, piccole pupille reattive,
atassia degli arti di tipo cerebellare, e perdita dell’emisensibilità
controlaterale (Caplan e Goodwin, 1982). Coloro che sopravvivono possono
sviluppare un mioclono
oculopalatale . La diagnosi può essere effettuato tramite meglio con
RM o TAC, o una combinazione di entrambe.
AICA (arteria cerebellare
antero inferiore)sindrome.
L'arteria cerebellare anteriore inferiore(AICA)
è un'arteria nel cervello che fornisce parte del cervelletto .
Esso deriva dalla arteria basilare a livello della giunzione tra
il midollo allungato ed i ponte nel tronco cerebrale . Si passa all'indietro da
distribuire alla parte anteriore della superficie inferiore del cervelletto,
anastomosi con il posteriore inferiore cerebellare ramo dell'arteria vertebrale
. Fornisce il quartiere inferiore anteriore del cervelletto.
Dà anche origine, nella maggior parte dei casi dell'arteria
labirintica; Tuttavia, in altri casi l'arteria labirintica può emergere come un
ramo della basilare. La sindrome AICA solito si presenta con vertigini e
sordità omolaterale unilaterale dalla labirintica ischemia dell'arteria. I
grandi ictus sono accompagnati da paralisi facciale omolaterale e atassia. E '
per frequenza il secondo più comune ictus del tronco cerebrale, circa il 10%
degli ictus colpiscono la PICA.
L'entità
di questo tratto è estremamente variabile. Sintomi simili a quelli della
malattia di Meniere (udito fluttuante, acufeni, vertigini) possono essere
causati anche da TIA in tale distribuzione (Lee e Cho, 2003). La
bilateralità delle fluttuazione uditive suggerisce una causa vascolare, ma la
maggior parte degli ictus da AICA si presentono con sintomi uditivi unilaterali. L'AICA
ha anche una origine molto variabile e può prendere origine dalla linea mediana
caudale del ponte. Gli ictus del territorio dell’AICA possono
presentare anche solo vertigini . La diagnosi si ottiene generalmente
tramite risonanza magnetica.
References
Lee H, Cho
YW. Auditory disturbance as a prodrome of anterior inferior cerebellar
artery infarction. J.
Neurol Neurosurg Psych 2003:74:1644-1648
Tab. I - Sintomi e
strutture nervose interessate nelle lesioni infartuali nel territorio
irrorato dalla P1CA e da AICA
Sintomi
Infarto nel
territorio
della PICA
Infarto nel
territorio
della AICA
Vertigine
Nn. vest., distretto
cereb. post-inf.
Distretto dorsolat.
b-pontino, flocculo, labirinto
Ipoacusia, acuf., vertigi-
ne
Assenti
Ipoacusia, acuf.,
vertigine
Segni di atas sia -
Presenti (tratto
cereb. ventrale)
Presenti (ped cereb.
medio).
Disfagia
I X
Assente
Disfonia
X
Emianest. fac- ciale
omolat
V
V
Paralisi del
Presente
Presente
Emi-ipoeste- sia
controlat
Presente (tratto
spino-tal.)
Presente (tratto
spino-tal.)
S di Bernard-Horner
Fibre simp.
disendent.
Fibre simp.
disendent.
Lesioni iperintense Pontine.
E
'comune incontrare le aree di maggiore segnale su T2 MRI nel ponte in persone
anziane con instabilità. Questi pazienti mostrano spesso i sintomi di
disequilibrio, disartria vocale e difficoltà alla deglutizione. (Kwa et
al, 1998). Secondo l'esperienza dell'autore, questi pazienti spesso
mostrano
nistagmo
di rimbalzo , che è una variante del nistagmo
evocato dallo sguardo.
Una
rara fonte di lesioni iperintense pontine è la mielinolisi
pontina centrale (vedi sopra). Questo è causato da rapide fluttuazioni
elettrolitico, solitamente nel contesto di un ricovero. L'individuo
mostrato sopra aveva un trapianto di fegato fatto. Dopo il trapianto di
fegato, è stato bene per un paio di giorni, ma poi è diventato a poco a poco in
coma. La sua risonanza magnetica in quel momento ha mostrato di sopra
l'immagine. Esame nove mesi più tardi rivelò un individuo ambulatoriale
con alcuni segni cerebellari lievi. Circa il 2% delle persone con
trapianto di fegato sviluppare mielinolisi pontina centrale.
Gli
individui con infarti della linea mediana pontina hanno di solito un normale ABR (Faught
e Oh, 1985).
REBOUND NISTAGMO oNISTAGMO DI
RIMBALZO
Il nistagmo
di rimbalzo
Re bound nystagmus is a primary position
nystagmus which is provoked by prolonged eccentric gaze holding.è associato
ad un nistagmo posizione primaria che è provocata da una sguardo prolungato in
posizione eccentrica. It appears after the eyes are
returned to primary position. Appare dopo che gli occhi ritornano alla
posizione primaria. Raramente
il NISTAGMO
DI RIMBALZO si
verifica in situazioni in cui non vi è malattia cerebellare evidente
There
are two methods of eliciting rebound.Ci sono due metodi per il nistagmo di rimbalzo.
The traditional method is to have the patient follow
ones finger to one side, hold gaze there for 10 seconds (with constant
encouragement by the examiner to keep looking), and then rapid return to
central gaze. Il metodo tradizionale è quello di avere il paziente segua
quelli dito da un lato, tenere lo sguardo lì per 10 secondi (con l'incoraggiamento
costante dall'esaminatore continuare a guardare), e poi un rapido ritorno allo
sguardo centrale. At that point, the examiner looks
for a nystagmus that beats away from the previous direction of gaze holding,
lasting for at least 5 beats. A quel punto, l'esaminatore cerca un
nistagmo che batte lontano dalla precedente direzione dello sguardo della
holding, della durata di almeno 5 battiti.
A more modern and sensitive method of eliciting rebound is to use
video-freznel goggles .Un metodo più
moderno e sensibile per evidenziare il nistagmo da rimbalzo è quello di utilizzare
gli
occhiali
video frenzel
. (as shown above). (Come indicato sopra). Otherwise the technique is similar. Altrimenti la
tecnica è simile. The video-frenzels make it much
easier to see small amounts of nystagmus. I video-frenzels rendono molto
più facile vedere piccole quantità di nistagmo.
Normal subject -- no significant nystagmus appears after 10
seconds of eccentric gaze holding. Soggetto normale - dopo 10 secondi
di sguardo eccentrico appare un nistagmo non significativo.
INormal subjects only rarely have rebound -- mainly after
very prolonged gaze-holding which is a different paradigm than discussed above
(Gordon et al, 1986; Shallo-Hoffmann, Schwarze et al. 1990; Suzuki 1991)
Soggetti normali solo raramente hanno un nistagmo da rimbalzo - soprattutto
dopo uno sguardo-holding molto prolungato che è un paradigma diverso da quello
discusso in precedenza (Gordon et al, 1986; Shallo-Hoffmann, Schwarze et al
1990; Suzuki
1991.)
Oggetto
anomala - sul pannello superiore (posizione), c'è lo sguardo evocata nistagmo
che batte a destra. When the eye returns to
center, there is a left-beating nystagmus. Quando l'occhio torna al
centro, c'è un nistagmo che batte a sinistro. Eye
velocity gradually declines during gaze holding to the right, and also decays
after return to center. Velocità Eye diminuisce gradualmente durante
lo sguardo tiene a destra, e anche decade dopo il ritorno al centro.
TC del soggetto a sinistra che mostra
i danni all'emisfero destro cerebellare. Image
courtesy of Dr. Immagine per
gentile concessione di Dr.Dario
Yacovino.Dario Yacovino
An abnormal amount of
rebound in the light, as shown above and below, consists of at least 3 beats of
clear nystagmus, with the slow-phases directed towards the previous position of
gaze.
Una quantità abnorme di rimbalzo alla luce, come mostrato sopra e sotto, è
costituito da almeno 3 battiti di evidente nistagmo, con fasi dirette lentamente
verso la posizione precedente dello sguardo. It must
reverse direction according to the direction of previous gaze. Si deve
invertire la direzione secondo la direzione dello sguardo precedente. When using the video-frenzel goggles, at least 5 beats
should be observed. Quando si utilizzano gli occhiali video Frenzel, si devono
essere osservate almeno 5 battiti.
Nistagmo Di Rimbalzo - un nistagmo che
batte a destra compare dopo 10 secondi di sguardo eccentrico a sinistra. This patient had a cerebellar disturbance. Questo
paziente aveva un disturbo cerebellare
Un Nistagmo Di Rimbalzo che batte a
sinistra compare dopo 10 secondi di sguardo eccentrico a destra.
Un nistagmo di
rimbalzo Re bound nystagmus is a primary position nystagmus which is provoked by
prolonged eccentric gaze holding.Rebound
after gaze holding for periods more prolonged than 30 sec, or for
eccentricities larger than about 45 deg is of uncertain significance as normal
subjects may exhibit rebound under such circumstances (Gordon et al, 1986).
dopo lo sguardo della holding per periodi più lunghi di 30 secondi, o per
eccentricità maggiore di circa 45 gradi è di importanza incerta in quanto i
soggetti normali possono presentare nistagmo dirimbalzo in tali
circostanze (Gordon et al, 1986). Vertical rebound
is rare but it can also occur. Il nistagmo di rimbalzo verticale
è raro, ma si può anche verificare.
Il nistagmo di
rimbalzo (Re bound nystagmus is a primary position nystagmus which is provoked by
prolonged eccentric gaze holding.Rebound) è quasi sempre patologico,
ed è principalmente legata a malattie del tronco cerebrale o del cervelletto
(Lin et al, 1999). Accordingly, if an unusually
large gaze-evoked nystagmus is observed, one should automatically look for
rebound nystagmus. Di conseguenza, se si osserva un grande nistagmo evocato
dalla direzione dello sguardo insolito, si dovrebbe cercare automaticamente un nistagmo
di rimbalzo. On the other hand, gaze-evoked
nystagmus without rebound is usually of little significance. D'altra
parte, un nistagmo evocato dalla direzione dello sguardo senza rimbalzo è
solitamente poco significativo. Rebound is always
associated with poor smooth pursuit , but the poor pursuit
does not inevitably mean that the person will have rebound. Il nistagmo di
rimbalzo (Rebound) è sempre associata a scarsa
inseguimento liscio
, ma il povero inseguimento non necessariamente significa che la persona avrà
rimbalzo.
Rebound nystagmus in patient with myotonic dystrophy -- type
II.
nistagmo di rimbalzo in pazienti con distrofia miotonica - tipo II.
Raramente il nistagmo di rimbalzo si
verifica in situazioni in cui non vi è malattia cerebellare evidente. Rarely rebound rarely occurs in situations where there is
no obvious cerebellar disease.Recentemente abbiamo trovato pazienti con distrofia
miotonica che hanno rimbalzo, probabilmente a causa di miotonia oculare. An example of this is shown above. Un esempio di
questo è mostrato sopra. (Driss et al, In press).
(Driss et al, in stampa). Persons with myotonia are
slow to relax previously contracted muscles. Le persone con miotonia
sono lenti a rilassare i muscoli contratti in precedenza. If this occured in the eye muscles, as others have
suggested (Versino et al, 2002) it might cause rebound nystagmus. Se
questo si è verificato nei muscoli oculari, come altri hanno suggerito (Versino
et al, 2002) potrebbe causare rimbalzo nistagmo.
Situations where
rebound nystagmus may occur Situazioni di eventuale rimbalzo
nistagmo
Pontine lesions (such
as MS or hypertension) Lesioni Pontine (come MS o ipertensione)
Cerebellar
degenerations (such as EA2) Degenerazioni cerebellari (come
EA2)
Congenital nystagmus Nistagmo
congenito
Normal persons after
prolonged gaze holding Persone normali, dopo lo sguardo prolungato
possesso
Myotonic dystrophy La
distrofia miotonica
Rebound using the video
goggles is usually a sensitive and specific sign of cerebellar disturbance. Il Nistagmo
di Rimbalzo evidenziabile con la video nistagmoscopia e/o grafia di solito
è un segno sensibile e specifico di disturbo cerebellare. Clinical situations in which rebound is commonly
encountered include MS involving the pons, typically a lesion of the middle
cerebellar peduncle, and ischemic pontine hypertensive lesions.
Rebound nystagmus is also a feature of EA2 ( Episodic ataxia, type 2 ). Situazioni
cliniche in cui rimbalzo è comunemente incontrato comprendono la SM quando
coinvolge il Ponte,è tipicamente una lesione del peduncolo cerebellare medio,
lesione Pontine
ischemica lesioni ipertesi. IL Rebound nistagmo è anche una caratteristica di
EA2 ( Atassia
episodica, tipo 2
). Rebound also can occur in congenital nystagmus , but it is not at all a
universal feature of CN.
Il Nistagmo di Rimbalzo (Rebound) ( può verificarsi anche nel nistagmo
congenito
, ma non è affatto una caratteristica universale di CN.
References: Riferimenti:
Ajroud-Driss S, Sufit R, Siddique T, Hain TC.
Ajroud-Driss S, Sufit R, Siddique T, TC Hain. Oculomotor
involvement in Myotonic Dystrophy Type 2. Submitted to Muscle/Nerve
Accepted 6/2008Coinvolgimento oculomotor in distrofia
miotonica di tipo 2. Presentata Muscle / Nerve accettati 6/2008
Gordon SE, Hain TC,
Zee, DS and Fetter M. (1986) Rebound nystagmus. Gordon SE,
Hain TC, Zee, DS e Fetter M. (1986) Rebound nistagmo. Soc. Soc. Neurosci.
Neurosci. Abstr., 12:1091. Abstr.,
12:1091.
Lin CY, Young YH:
Clinical significance of rebound nystagmus. Lin CY, Giovane YH:
significato clinico di rimbalzo nistagmo. Laryngoscope.
Laringoscopio. 1999 Nov; 1999 Novembre; 109(11): 1803-5. 109 (11): 1803-5.
Shallo-Hoffmann J, Schwarze H, Simonsz HJ et al: A reexamination of
end-point and rebound nystagmus in normals. Invest Ophthalmol Vis Sci. 1990 Feb; Shallo-Hoffmann J, Schwarze H,
Simonsz HJ et al:. Un
riesame di end-point e rimbalzo nistagmo nei soggetti normali .
Invest Ophthalmol Vis Sci 1990 Feb; 31(2): 388-92. 31 (2): 388-92.
Suzuki T.: Rebound
nystagmus in normal subjects. Suzuki T.: Rebound nistagmo nei
soggetti normali. Nippon Ganka Gakkai Zasshi.
Nippon Ganka Gakkai Zasshi. 1991 Sep;
1991 Settembre; 95(9): 878-82. 95 (9):
878-82.
Versino M, Colnaghi S,
Sandrini G et al: Ocular motor myotonic phenomenon in myotonic dystrophy. Versino M,
S Colnaghi, Sandrini G et al: motore oculare fenomeno miotonica nella
distrofia miotonica. Ann NY Acad Sci.
Ann NY Acad Sci. 2002 Apr;956:401-4.
2002 Aprile; 956:401-4.
LaCentral pontine myelinolysis is a rare cause of damage
to the brainstem. Mielinolisi pontina centrale è una rara causa di un danno al tronco
encefalico.First described by Adams et al. Prima descritto da Adams et al.in 1959 in their chronic alcoholic patients, it has now
been described in the malnourished, the chronically debilitated, the renal, the
hepatic and the transplant patient among others.
nel 1959 a loro pazienti cronici alcoliche, è stato ora descritto nel
malnutriti, cronicamente debilitati, renale, epatica e il paziente trapianto
tra gli altri.CPM is caused by rapid
fluctuations in electrolyte status, usually in the context of a
hospitalization. CPM è causata da rapide
fluttuazioni in stato di elettrolita, generalmente nel contesto di un ricovero.About 2% of persons with liver transplant develop
central pontine myelinolysis. Circa il 2% delle
persone con trapianto di fegato sviluppare mielinolisi pontina centrale.Some cases of CPM appear to be triggered solely by
alcohol withdrawall (Yoon et al, 2008) Alcuni
casi di CPM sembrano essere innescato solo dall'alcol withdrawall (Yoon et al,
2008)
There is an
extensive literature concerning CPM, with more than 400 publications having
this in the title in Pubmed as of 5-2009. Esiste una vasta letteratura in materia CPM, con più
di 400 pubblicazioni avere questo nel titolo in Pubmed come di 5-2.009.
According to Lempl, as of 2006, 174 cases of CPM have
been reported in alcoholics since 1986, which is equivalent to an incidence of
39.4%.
Secondo Lempl, a partire dal 2006, 174 casi di CPM sono stati riportati negli alcolisti
dal 1986, il che equivale ad una incidenza del 39,4%.Thus chronic alcoholism
is still the most common underlying condition of CPM patients.Likewise, 95
cases of CPM following the correction of hyponatremia have been documented
since 1986 (21.5%). Così l'alcolismo cronico è
ancora la condizione di base più comune di CPM patients. Likewise, 95 casi di
CPM successivi alla correzione di iponatremia sono stati documentati dal 1986
(21,5%).The third largest group of CPM
cases are liver transplant patients (17.4%), with the development of CPM being
attributed to the immunosuppressive agent cyclosporine in particular. Il terzo gruppo di casi di CPM sono pazienti sottoposti a
trapianto di fegato (17,4%), con lo sviluppo di CPM essere attribuito alla ciclosporina
agente immunosoppressivo in particolare.
Diagnosis Diagnosi
Pathologically, it is defined as a symmetric area of
myelin disruption in the center of the basis pontis, although similar symmetric
lesions have also been described occurring with CPM as well as independently in
other brain areas (extrapontine myelinolysis or EPM) including the cerebellar
and neocortical white/gray junctional areas, thalamus and striatum. Patologicamente, essa è
definita come una zona simmetrica di interruzione mielina nel centro della
pontis base, anche se le lesioni simmetriche simili sono anche stati descritti
verificano con CPM e indipendentemente in altre aree cerebrali (mielinolisi extrapontina
o EPM) compreso il cerebellare e neocorticale bianco / grigio giunzionale aree,
talamo e striato.
Clinically, patients often are unable to move their
eyes during their acute period, may be "locked in" -- with
quadraparesis, and have speech disturbance. Clinicamente, i pazienti
spesso sono in grado di muovere gli occhi durante il loro periodo di acuta, può
essere "bloccata in" - con tetraparesi, e hanno disturbi del
linguaggio.
Case example 1: osmotic syndrome. Esempio Caso 1: la sindrome
osmotica.a patient presented to the clinic complaining of chronic
dizziness and imbalance. un paziente ha
presentato al lamentarsi clinica di capogiro cronico e squilibrio.During a hospitalization for pancreatitis, he became
hyponatremic. Nel corso di un ricovero per la
pancreatite, è diventato iponatriemia.An
MRI scan showed "poorly defined high T2 signal within the central
pons". Una risonanza magnetica ha mostrato
"il segnale ad alta T2 mal definita all'interno del ponte centrale".Physical examination revealed slowing of upgoing
saccades and positional nystagmus. L'esame
fisico ha rivelato rallentamento delle saccadi che sale e nistagmo posizionale.
Case 2: Liver transplant. Caso 2: Il trapianto di
fegato.The individual shown below had a liver transplant done. L'individuo indicato di seguito ha avuto un trapianto di
fegato fatto.After the liver transplant,
he was fine for a couple of days but then gradually became comatose. Dopo il trapianto di fegato, è stato bene per un paio di
giorni, ma poi è diventato a poco a poco in coma.His MRI at that time showed the picture above. La sua risonanza magnetica in quel momento ha mostrato sopra
l'immagine.Examination nine months later
revealed an ambulatory individual with some mild cerebellar signs. Esame nove mesi più tardi ha rivelato un individuo
deambulatorio con alcuni segni cerebellari miti.
MRI scan of
person with central pontine myelinolysis. Risonanza magnetica di
paziente con mielinolisi pontina centrale.Saggital view The dark
area inside the circle is the region of damage. Vista sagittale L'area scura all'interno del cerchio è la
regione del danno.
MRI scan of
person with central pontine myelinolysis, axial view. Risonanza magnetica di
persona con Mielinolisi pontina centrale, vista assiale.Note the "I"
shaped area in the center of the pons. Nota l
'"io" a forma di zona al centro del ponte.
Mechanism Meccanismo
According to
Kumar and associates (2006), central pontine myelinolysis (CPM) can be regarded
as one of the demyelinating syndromes. Secondo Kumar e colleghi (2006), mielinolisi pontine
centrale (CPM) può essere considerato come una delle sindromi demielinizzanti.
Possible
mechanisms include a hyperosmotically induced demyelination process resulting
from rapid intracellular/ extracellular to intravascular water shifts producing
relative glial dehydration and myelin degradation and/or oligodendroglial
apoptosis.
I possibili meccanismi comprendono un processo di demielinizzazione iperosmotica
indotto derivante dal rapido intracellulare / extracellulare a turni acqua
intravascolari producono relativa gliali disidratazione e mielina degrado e / o
apoptosi oligodendrogliali.CPM is associated with the immunosuppressant agent
cyclosporine, which is mainly used for transplants. CPM è associato con la ciclosporina agente
immunosoppressore, che viene utilizzato principalmente per il trapianto.
Treatment: Trattamento:
Avoidance of CPM is dependent upon recognizing those
patients with conditions pre-disposing them to osmotic myelinolysis and then
moderating the rate of normalization of the electrolyte imbalance. Evitare di CPM dipende
riconoscere quei pazienti che presentano condizioni predisponenti per
mielinolisi osmotica e quindi moderare la velocità di normalizzazione del
squilibrio elettrolitico.Treatment after the CPM has developed is mainly
supportive, but successful treatment with a variety of agents including
plasmapheresis (Grimaldi et al, 2005) and TRH (Zein et al, 2006) have been
reported. Il trattamento dopo il CPM si è
sviluppato è principalmente di supporto, ma il trattamento di successo con una
varietà di agenti compresa la plasmaferesi (Grimaldi et al, 2005) e TRH (Zein
et al, 2006) sono stati segnalati.It is
difficult to see the rationale for these treatments. E 'difficile vedere la logica per questi trattamenti.
Most patients recover without any special treatment. La maggior parte dei pazienti
guarisce senza alcun trattamento speciale.
For symptoms that persist after several months, we
sometimes attempt to manage them for "central dizziness", and combine
physical therapy (if appropriate) use medications intended to affect the
brain such as gabapentin, or 3-4 DAP. Per i sintomi che persistono dopo diversi mesi, a
volte cerchiamo di gestirli per "vertigini centrali", e uniamo la terapia fisica (se del caso) di utilizzare i farmaci destinati ad influenzare il cervello come il
gabapentin o 3-4 DAP.
References:
Riferimenti:
Grimaldi,
D., F. Cavalleri, et al. Grimaldi, D., F.
Cavalleri, et al.(2005). (2005)."Plasmapheresis
improves the outcome of central pontine myelinolysis." "La plasmaferesi migliora il risultato di
mielinolisi pontina centrale."J
Neurol 252(6): 734-5. J Neurol 252 (6):
734-5.
Kumar, S., M. Fowler, et al. Kumar, S., M. Fowler, et al.(2006).(2006)."Central pontine myelinolysis, an
update." "Mielinolisi pontina
centrale, un aggiornamento."Neurol
Res 28(3): 360-6. Neurol Res 28 (3): 360-6.
Lampl,
C. and K. Yazdi (2002). Lampl, C. e K.
Yazdi (2002)."Central pontine
myelinolysis." "Mielinolisi
pontina centrale."Eur Neurol
47(1): 3-10. Eur Neurol 47 (1): 3-10.
Yoon, B., YS Shim, et al. Yoon, B., YS Shim, et al.(2008).(2008)."Central
pontine and extrapontine myelinolysis after alcohol withdrawal." "Pontina centrale e Mielinolisi extrapontina dopo
il ritiro di alcol."Alcohol
Alcohol 43(6): 647-9. Alcol 43 (6): 647-9.
Zein,
EF, SE Karaa, et al. Zein, EF, SE Karaa, et
al.(2006). (2006)."Treatment
of central pontine myelinolysis with thyrotropin-releasing hormone." "Il trattamento di pontina centrale mielinolisi
con l'ormone rilasciante la tireotropina."Presse Med 35(4 Pt 1): 618-20. Presse Med 35 (4 Pt 1): 618-20.
· Contributi originali
Infarto nel territorio di Anteriore
Inferiore Artery Cerebellare
1.Dal Dipartimento di Neurologia (HL, E.-JC, H.-AY) e il Brain
Research Institute (HL, H.-AY, I.-SC, S.-RL), Keimyung University School of
Medicine, Daegu, Corea del Sud; e il Dipartimento di Neurologia (JSK,
J.-YS), Seoul National University College of Medicine, Seoul National
Università Bundang Hospital, Seongnam, Corea del Sud.
1.Corrispondenza di Hyung Lee, MD, Dipartimento di Neurologia,
Keimyung University School of Medicine, 194 Dongsan dong, Daegu, 700-712 Corea
del Sud.E-mail hlee @ dsmc.or.kr
Contesto
e lo Scopo Per definire lo spettro dettagliata della disfunzione
Audiovestibolare in anteriore inferiore arteria cerebellare territorio
miocardico.
Metodi- oltre
8,5 anni, abbiamo identificato prospetticamente 82 pazienti consecutivi con
infarto anteriore inferiore territorio arteria cerebellare diagnosticata da
risonanza magnetica. Ogni paziente ha completato un questionario
standardizzato audiovestibulare ed ha subito una valutazione neuro-otologica,
compresi i test calorico bitermico e audiogramma tono puro.
Risultati Tutti
tranne 2 (80 di 82 [98%]) pazienti avevano vertigini prolungata acuto e
disfunzione vestibolare periferico, centrale, o di origine combinato. Il
modello più comune di disfunzione audiovestibular è stata la perdita combinata
di funzione uditiva e vestibolare (n = 49 [60%]). Una perdita selettiva di
vestibolare (n = 4 [5%]) o cocleare (n = 3 [4%]) funzione è stata osservata
raramente. Potremmo classificare anteriore inferiore cerebellare
territorio dell'arteria infarto in 7 sottogruppi in base ai modelli di
presentazioni otoneurologici: (1) acuta vertigini prolungata con perdita
audiovestibular (n = 35); (2) acuta vertigine prolungato con perdita
audiovestibulari preceduto da un episodio (s) di vertigine transitoria /
disturbo uditivo entro 1 mese prima del miocardio (n = 13); (3) acuta
prolungata vertigini e perdita uditiva isolato senza perdita vestibolare (n =
3); (4) acuta prolungata vertigini e isolato perdita vestibolare senza
perdita uditiva (n = 4); (5) acuta prolungata vertigine, ma senza perdita
documentata audiovestibulare (n = 24); (6) acuta prolungata vertigine e
perdita audiovestibulari isolato, senza altri sintomi neurologici / segni (n =
1); e (7) sintomi non vestibolari con normale funzione audiovestibulari (n
= 2).
Infarto In
conclusione, nella parte anteriore inferiore cerebellare
territorio dell'arteria può presentare con un ampio spettro di disfunzioni
audiovestibulari. A differenza di una causa virale, la disfunzione
labirintica di una causa vascolare di solito porta alla perdita combinata di
entrambe le funzioni uditive e vestibolari.
Nel infarto che
coinvolgono la distribuzione del anteriore inferiore arteria cerebellare
(AICA), vertigini è di solito associata con altri sintomi neurologici o segni
come la perdita dell'udito, debolezza facciale, perdita di sensibilità del
viso, attraversato perdita di sensibilità, la sindrome di Horner, andatura
atassia, e degli arti atassia . 1-3 Adams 1 è stato il primo che
descrisse completamente la sindrome associata con AICA occlusione. Nel suo
paziente, sintomi neuro-otologica quali vertigini, acufeni e sordità bilaterale
sono stati i primi sintomi. Le relazioni successive sul AICA miocardico si
sono concentrati sui risultati del tronco cerebrale e cerebellare con poca
attenzione alla disfunzione otoneurologici associato. 2,4-7Prima del 2000, pochi
studi 8,9 avevano
accuratamente studiato i disturbi audiovestibular che si verificano con AICA
infarto.
AICA di solito
nasce dal terzo caudale dell'arteria basilare e fornisce l'orecchio interno,
pons laterali, mezzo peduncolo cerebellare, e anteriore del cervelletto
inferiore, compreso il flocculo. 2,5 Perché AICA è
un'arteria importante per apporto vascolare alle strutture vestibolari
periferici e centrali e la sua occlusione comunemente provoca vertigini sia di
origine centrale o periferica, abbiamo considerato che l'analisi dei modelli di
perdita vestibolare può far luce sul meccanismo di vertigine che si verificano
in una compromissione vascolare all'interno del circolo posteriore.
Anche se recenti
studi 10-17 hanno sottolineato
che la perdita audiovestibular è un segnale importante per la diagnosi di
infarto AICA, spettro dettagliato di disfunzioni audiovestibular non è stato
studiato sistematicamente in AICA territorio infarto. Quindi, abbiamo
cercato di indagare i modelli di disfunzione audiovestibular in un'ampia serie
di pazienti consecutivi con infarto territorio AICA e di considerare la sua
implicazione clinica.
Tra il gennaio 2000
e luglio 2008, sono stati identificati 90 pazienti consecutivi dal registro
acuto corsa a 2 ospedali universitari in Corea, che hanno avuto un infarto
acuto nella distribuzione del AICA. Abbiamo determinato il territorio AICA
utilizzando i diagrammi anatomici di Amarenco e Hauw 3 e la diagnosi
dell'infarto AICA è stata fatta quando le lesioni MRI coinvolti almeno una
delle seguenti strutture anatomiche: mezzo peduncolo cerebellare, laterale zona
pontine inferiore, o antero emisfero cerebellare . Il tipico AICA
territorio pontino lesione è come un triangolo con una base anterolaterale e un
apice rivolto verso il quarto ventricolo tra il peduncolo cerebellare medio e
regione pontina anterolaterale. Dopo aver escluso 8 pazienti con
incompleta valutazione audiovestibular, 82 pazienti sono stati infine
selezionati per questo studio. In ogni paziente, i test diagnostici sono
stati eseguiti per determinare i fattori di rischio di ictus. L'età media
dei pazienti era di 64,9 ± 15,3 anni, con un range da 23 a 93 anni. Ogni
paziente ha completato un questionario standardizzato vertigini che comprendeva
una descrizione dettagliata sui disturbi audiovestibular acuti e sottoposto ad
una valutazione neuro-otologica svolto dagli autori (HL e JSK).
Vertigo è stata
definita come una illusione filatura dell'ambiente o pazienti stessi.Test
calorico è stato eseguito a 30 ° C e 44 ° freddo C irrigazioni calde ciascun
orecchio per 20 secondi. Canal paresi (CP) è stato calcolato utilizzando
la formula Jongkees. I dettagli relativi alle prove audiovestibular sono
stati precedentemente pubblicati. 3,18 Abbiamo definito la
perdita uditiva di una causa vascolare come segue: (1) i pazienti hanno notato
un definitivo declino dell'udito durante l'attacco di AICA miocardico; e
(2) tono puro audiogramma anche documentato la perdita dell'udito
neurosensoriale (SNHL). La diagnosi di perdita vestibolare di una causa
vascolare era basata sui seguenti criteri: (1) il paziente aveva prolungato
acuta (> 24 ore) vertigine al momento di AICA miocardico; e (2)
stimolazione calorica standardizzata mostrato una ridotta risposta> 25% nel
lato della lesione.Disfunzione vestibolare centrale è stata definita quando:
(1) il paziente aveva prolungato (> 24 ore) vertigine acuta al momento della
AICA miocardico; (2) stimolazione calorica standardizzata mostrava normale
risposta calorico; e (3) pazienti hanno mostrato almeno uno dei seguenti
segni, incluse quelle asimmetriche di inseguimento o nistagmo optocinetico,
sguardo-evocato nistagmo bidirezionale o alterata modulazione della risposta
vestibolare utilizzando input visivo.
Valutazioni
otoneurologici sono stati in gran parte eseguiti durante il periodo di acuta (0
a 43 giorni, intervallo di 6,7 giorni per la valutazione vestibolare e 5,3
giorni per la valutazione uditiva significare). Nella maggior parte dei
pazienti, la risonanza magnetica, tra cui l'imaging pesato in diffusione e MR
angiografia (MRA), è stata eseguita durante il periodo di acuta (0 a 30 giorni,
significano intervalli, 4.1 giorni).
Le analisi
statistiche sono state eseguite utilizzando il programma SPSS (versione 12.0,
SPSS, Chicago, Illinois). χ 2 test è stato
utilizzato per confrontare il risultato di risultati MRA nei pazienti con e
senza perdita audiovestibular prodromica. I risultati MRA nei pazienti con
e senza più infarti circolazione posteriore sono stati confrontati con il test
χ 2. La significatività è stata assunta ad un valore di
P <0,05.
Tutti gli
esperimenti hanno seguito i principi della Dichiarazione di Helsinki e consensi
informati sono stati ottenuti dopo la natura e le possibili conseguenze dello
studio erano stati spiegati ai partecipanti. Poiché il presente studio ha
incluso tutti i pazienti consecutivi durante il periodo di ricerca, 23 pazienti
precedentemente riportato 3,10-12,16,17 sono stati
inclusi, ma le nuove informazioni si aggiunge in questo rapporto.
Tutti erano attenti
e orientato al momento del ricovero. Nella maggior parte dei pazienti (80
su 82 [98%]), vertigine acuta spontanea prolungata (> 24 ore) con nausea /
vomito è stata la presentazione o il sintomo principale. Il nistagmo
spontaneo era prevalentemente orizzontale e 55 (67%) pazienti hanno mostrato
nistagmo spontaneo battendo distanti (84% [46 55]) o verso (11% [6 di 55]) il
lato della lesione. Gli altri 3 hanno mostrato altalena, ottimista, o puro
nistagmo torsionale. Asimmetrico bidirezionale nistagmo sguardo-evocato è
stato trovato anche in 35 pazienti. Altri risultati inclusi arto dismetria
(n = 55 [67%]), andatura atassia (n = 52 [63%]), perdita di sensibilità
facciale (n = 23 [28%]), debolezza facciale (n = 23 [21%] ), perdita di
sensibilità corporea (n = 5 [6%]), la sindrome di Horner (n = 3 [4%]),
disartria (n = 3 [4%]), occhio limitazione del movimento (n = 2 [2%]) e
debolezza degli arti (n = 2 [2%]). La sindrome completa AICA descritto da
Adams è stato trovato in solo 2 (2%) pazienti in cui i classici sintomi pontine
come la perdita del viso sensoriale e debolezza, attraversò segno sensoriali, e
la sindrome di Horner sviluppato oltre a vertigini prolungata, perdita di
udito, e cerebellare segni. Il fattore di rischio più comune era
l'ipertensione (66%) seguita da diabete mellito (40%), abitudine al fumo (27%),
una storia di ictus (23%), la fibrillazione atriale (7%), e le malattie
cardiache (6%) . Almeno 2 fattori di rischio vascolare sono stati trovati
in 40 (49%), mentre nessun fattore di rischio identificabile era presente in 8
(10%) pazienti.
Caratteristiche di perdita dei Audiovestibular
C'erano 7
sottogruppi di AICA territorio infarto secondo i modelli di presentazioni
otoneurologici (Tabella 1): (1) 35 pazienti ha
presentato con vertigini prolungata e ha avuto una perdita audiovestibular
acuta (cioè, CP e SNHL); (2) 13 pazienti avevano un episodio (s) di
vertigine transitoria, perdita dell'udito, e / o tinnito entro 1 mese prima
dell'infarto oltre a vertigini prolungata e perdita audiovestibular acuta come
nel gruppo 1; (3) 3 pazienti presentato con vertigini prolungata e aveva
insorgenza acuta di SNHL isolato senza CP; (4) 4 pazienti hanno presentato
con vertigini prolungata e isolato CP senza SNHL; (5) 24 pazienti ha
presentato con vertigini, ma non aveva CP di accompagnamento o SNHL; (6)
un paziente ha presentato con vertigini prolungata e isolato perdita
audiovestibular senza altri sintomi o segni neurologici; e (7) 2 pazienti
presentati con insorgenza improvvisa di perdita di sensibilità o di atassia e
disartria andatura senza vertigini o perdita audiovestibular. Le frequenze
e le caratteristiche dei disturbi audiovestibular sono riassunti nella Tabella 2.
Tabella 2. Le frequenze delle Audiovestibular Disfunzioni in 82
pazienti con AICA Territorio Infarto
Tutti tranne 2 (80
di 82 [98%]) dei pazienti ha avuto una disfunzione vestibolare periferico,
centrale, o di origine combinato. La maggior parte (96% [79 di 82]) dei
pazienti ha mostrato che accompagna motorie oculari o vestibolari segni
centrali che sono stati caratterizzati da asimmetrica esercizio regolare e
nistagmo optocinetico, bidirezionale nistagmo sguardo-evocato, e modulazione
anormale del riflesso vestibolo-oculare utilizzando input visivo. Circa il
70% (56 di 82) dei pazienti ha mostrato due CP (53 di 82 [65%]) o SNHL (52 di
82 [63%]). La maggior parte di essi (49 56 [88%]) avevano combinato
vestibolococleare erettile (cioè, CP e SNHL), mentre solo 4 (7%) era isolato CP
senza SNHL e 3 (5%) avevano isolato SNHL senza CP. CP era unilaterale e
era sul lato di infarto dimostrato alla risonanza magnetica. Al contrario,
circa il 30% (26 su 82) dei pazienti non ha mostrato alcuna evidenza di
disfunzione audiovestibular, anche se la maggior parte di loro (92% [24 di 26])
vertigini aveva anche prolungata. Audiogramma tonale rilevato unilaterale
(n = 50) o bilaterale (n = 2) SNHL nel periodo acuta. Il SNHL unilaterale
era sul lato di infarto dimostrato alla risonanza magnetica. Dei 2
pazienti con SNHL bilaterale, uno aveva improvvisa perdita di udito bilaterale
di grado moderato alla presentazione iniziale e l'altra mostrava solo lato
sinistro sordità profonda al momento del ricovero, ma successivamente
sviluppato ipoacusia diritto di grado moderato durante l'ospedalizzazione. Nessuno
dei pazienti aveva una storia di un'eccessiva esposizione al rumore, trauma
cranico, la meningite, farmaci ototossici o la sifilide.Sette pazienti avevano
una storia di malattia dell'orecchio medio, ma c'era un chiaro aggravamento
della perdita dell'udito (SNHL su audiogramma tono puro) al momento di infarto. MRI
e risultati audiovestibular dei pazienti rappresentativi dei gruppi 1 e 3 sono
mostrati in figure 1 e 2⇓rispettivamente.
Figura 1. RM e risultati audiovestibular
in un paziente con infarto territorio AICA e perdita audiovestibular (Gruppo
1). A, risonanza magnetica assiale pesata in diffusione dimostra infarto
acuto che coinvolgono il diritto peduncolo cerebellare medio e anteriore destra
emisfero cerebellare. B, audiogramma tono puro rivela grave grado di
perdita dell'udito a destra. C, registrazioni video-oculografiche di prove
caloriche bitermico fornisce destra paresi del canale (89%). Vmax,
velocità massima di lenta fase del nistagmo; PTA, audiogramma tono puro.
Figura 2. RM e risultati audiovestibular
in un paziente con infarto territorio AICA e isolato la perdita uditiva senza
perdita vestibolare (Gruppo 3).A, RM assiali pesate in T2 dimostra infarto
acuto che coinvolge il ponte dorsolaterale sinistra. B, Un PTA iniziale
rivela perdita dell'udito lieve sul lato sinistro. C, PTA Follow-up rivela
2 settimane più tardi i livelli normali udito su entrambi i lati.D,
registrazioni elettro-oculografiche di prove caloriche bitermico fornisce
risposte normali su entrambi i lati. Vmax, velocità massima di lenta fase
del nistagmo; PTA, audiogramma tono puro.
Topografia di infarti associati alla perdita Audiovestibular e
risultati MRA
Il peduncolo
cerebellare medio è stato influenzato in 68 (83%), anteriore cervelletto inferiore
a 38 (46%), e ponte inferiore laterali in 36 (44%) pazienti. AICA infarto
unilaterale in 79 (96%) e bilaterale in soli 3 (4%) pazienti che hanno mostrato
lesioni in entrambi i peduncoli cerebellari medi. Completa infarto AICA
che coinvolge il peduncolo cerebellare medio, ponte laterali, anteriori e
cervelletto inferiore è stato trovato in solo 13 (16%) pazienti. Secondo i
risultati di immagini pesata in diffusione, sono state identificate 3 categorie
di infarto. In primo luogo, isolati infarti AICA (n = 55) sono stati
limitati al peduncolo cerebellare medio (n = 44), dorsolaterale pontino
tegmentum (n = 26), o anteriore del cervelletto inferiore (n = 27). In
secondo luogo, AICA e altri infarti cerebellari (n = 14) AICA coinvolto più
posteriore inferiore arteria cerebellare o territorio arteria cerebellare
superiore.AICA infarto era unilaterale in entrambi i gruppi. Terzo, AICA
più infarti multipli (n = 13) hanno mostrato infarti multipli nella
circolazione posteriore territorio oltre a AICA miocardico. In 16 pazienti
(20%), MRA divulgate focale (ovvero, medio o inferiore parte vicino all'origine
AICA) o stenosi diffusa dell'arteria basilare. Trenta pazienti (37%) hanno
mostrato un restringimento diffuso o segmentale dell'arteria vertebrale ipsi- o
controlaterale alla infarto AICA. Un altro 41 (50%) pazienti hanno
mostrato normale dell'arteria vertebro-basilare su MRA. I pazienti con
disturbi audiovestibular prodromici hanno mostrato 5 volte più alta prevalenza
di una grave malattia dell'arteria occlusiva basilare rispetto ai pazienti
senza disturbi prodromica audiovestibular (62% contro 13%, p <0.001;Tabella 3). I pazienti con stenosi
basilare anche tendevano ad avere più infarti circolazione posteriore rispetto
ai pazienti senza (5 di 16 [31%] vs 8 di 66 [12%], p <0.05). Inoltre,
una grave malattia dell'arteria basilare occlusiva era più comunemente
osservata nei pazienti con AICA e molteplici infarti circolazione posteriore (6
di 13 [46%]) rispetto ai pazienti con AICA isolata o AICA più altri infarti
cerebellari (10 di 69 [14%], P <0,017).
Per quanto a nostra
conoscenza, questo è di gran lunga la più grande serie di AICA infarto
focalizzato sulle disfunzioni audiovestibular. Nella nostra serie, la
quasi totalità (98%) pazienti con infarto territorio AICA presentato con
insorgenza acuta di prolungata (> 24 ore) vertigini e aveva una disfunzione
vestibolare periferico, centrale, o di origine combinato. Il modello più
comune di disfunzione vestibolare nella nostra serie era una combinazione di
periferica (cioè, CP unilaterale) e Central Motor oculare o segni vestibolari
(cioè, in modo asimmetrico alterata esercizio regolare, bidirezionale nistagmo
sguardo-evocato, o la modulazione alterata delle risposte vestibolari con input
visivo) che si osserva in circa il 65% (53 su 82) dei pazienti. Questi
risultati possono essere spiegati dal fatto che AICA fornisce costantemente le
strutture vestibolari periferici come l'orecchio interno e nervo
vestibolococleare oltre alle strutture vestibolari centrali. 2,5 Di conseguenza, a
differenza di altro territorio dell'arteria cerebellare miocardico, infarto
completa AICA solito si traduce in danni combinati vestibolari periferiche e
centrali oltre alla perdita, debolezza facciale, degli arti e perdita
sensoriale facciale, andatura atassia e dismetria cerebellare dell'udito. A
causa di ischemia eventuali strutture fornite da AICA può portare a vertigini,
una conclusione definitiva sul sito (s) responsabile per la vertigine
prolungato sembra difficile nel singolo paziente con AICA infarto.Tuttavia, 53
(65%) pazienti con infarto AICA avevano una debolezza unilaterale di
stimolazione calorica, suggerendo che la vertigine era dalla disfunzione della
struttura vestibolare periferica, almeno in parte. D'altra parte, 27
pazienti (33%) hanno mostrato una normale risposta calorico, indicando che la
vertigine può essere il risultato di ischemia alle strutture vestibolari
centrali in questi pazienti.Nel complesso, i nostri risultati hanno dimostrato
che le vertigini prolungata AICA infarto per lo più risultati da ischemia sia
alle strutture vestibolari periferici e centrali.
Nella nostra serie,
il 60% (49 su 82) dei pazienti con AICA infarto mostrava insorgenza acuta di
perdita audiovestibular caratterizzato da CP e SNHL, che è in accordo con le
precedenti relazioni che la perdita audiovestibular è un segnale importante per
la diagnosi di AICA infarto. 3,12-16,17 Questo risultato può
essere spiegato da (1) l'arteria uditivo interno, l'arteria principale per
vascolarizzazione all'orecchio interno, di solito proviene da AICA; e (2)
l'orecchio interno è particolarmente sensibile all'ischemia transiente a causa
del suo fabbisogno energetico e alte mancanza di adeguato apporto di sangue
collaterale. 3,8-10,16-18A differenza di una
precedente relazione 2 che circa l'80% dei
pazienti con AICA miocardico ha mostrato sintomi o segni indicativi di disfunzione
pontino laterale, debolezza facciale o perdita della sensibilità incrociate
suggerendo disfunzione pontino è stato trovato nel solo il 28% (23 su 82) dei
pazienti della nostra serie, suggerendo che anche se i segni pontine sono le
caratteristiche principali che differenziano AICA infarto da un più comuni
patologie benigne che coinvolgono l'orecchio interno, è meno comune di quanto
si pensasse.
È noto che le
variazioni anatomiche sono comuni nella vascolatura cerebellare.Anche in
persone normali, l'AICA può dominare in un lato, mentre la parte posteriore
inferiore dell'arteria cerebellare fornisce principalmente cervelletto
inferiore nell'altro lato. A volte, sia la AICA o posteriore inferiore
arteria cerebellare è assente e un'arteria fornisce i soliti territori di
entrambe le arterie. Raramente, una posteriore inferiore arteria
cerebellare può irrigare entrambi i lati del cervelletto inferiore. Purtroppo,
solo alcuni dei nostri pazienti sottoposti angiografia convenzionale e la
patologia esatta di posteriore inferiore dell'arteria cerebellare è oltre la
risoluzione di MRA.
E 'interessante
confrontare i risultati MRA nei pazienti con disturbi audiovestibular
prodromici (definiti come episodio [s] di vertigine transitoria, perdita
dell'udito, e / o tinnito entro 1 mese prima del miocardio) con quelli dei
pazienti senza disturbi prodromica audiovestibular; focale o stenosi
diffusa dell'arteria basilare vicino all'origine di AICA era più comune nei
pazienti con disturbi prodromica audiovestibular (62% contro 13%, p = 0,000). Questa
scoperta potrebbe spiegare l'elevata incidenza di sintomi prodromici nel gruppo
di pazienti con compromissione dell'arteria basilare su MRA. Colpi
territoriali della AICA sono stati associati con la malattia dell'arteria
basilare ramo occlusiva. 2,5,6 Poiché la maggior
parte dei nostri pazienti con disturbi audiovestibular prodromici avevano
evidenza di una focale o segmento diffusa di riduzione del flusso sanguigno
nell'arteria basilare vicino all'origine AICA, una placca ateromasica
all'interno dell'arteria basilare può essere esteso in ostia AICA. Con
questo meccanismo, riduzione del flusso di sangue nel interessata AICA potrebbe
causare uno episodio transitorio di ischemia selettiva per l'orecchio interno,
con conseguente isolato disturbo prodromica audiovestibular, o di danni
permanenti alle aree diffuse che coinvolgono il peduncolo cerebellare medio,
ponte laterali, e anteriore del cervelletto dando luogo a vertigini prolungata
e perdita dell'udito, oltre ad altri sintomi centrali e segni. Nella
nostra serie, la maggior parte dei pazienti (45 su 55 [82%]), con isolati AICA
miocardio ha mostrato normale MRA e grave malattia dell'arteria basilare
occlusiva era più comune nei pazienti con infarti circolazione posteriore,
oltre a AICA infarto, in linea con la precedente relazione 2 che isolati infarti
AICA sono di solito causati da basilare malattia occlusiva ramo, mentre gli
infarti che si estendono oltre il territorio AICA sono per lo più a causa di
malattia dell'arteria basilare occlusiva.
I nostri risultati
hanno implicazioni pratiche. Quando i pazienti con fattori di rischio per
l'ictus sviluppati insorgenza acuta di isolato vertigini prolungata senza
accompagnamento perdita dell'udito o altri sintomi neurologici, danno ischemico
al labirinto vestibolare superiore a causa anteriore arteria vestibolare
infarto è ragionevolmente sospettato perché il lume anteriore dell'arteria
vestibolare è piccola e ha poco circoli collaterali. 9,19,20 Un rapporto
precedente 20 anche sostenuto
questa ipotesi perché circa il 50% dei pazienti con isolati vertigine episodica
di una causa vascolare (cioè, insufficienza vertebro-basilare) aveva CP
unilaterali, che è comunemente localizzata all'orecchio interno (cioè,
labirinto vestibolare superiore). Tuttavia, la nostra scoperta non ha
sostenuto questa ipotesi perché solo 4 (5%) pazienti hanno mostrato isolato
coinvolgimento labirintico vestibolare al momento della AICA infarto. Quindi,
anche se isolato anteriore vestibolare arteria infarto può essere servito come
un meccanismo di isolato vertigini vascolare, l'incidenza sarebbe basso. Isolata
coinvolgimento della coclea è anche manifestazione rara di AICA infarto nella
nostra serie, che è stata osservata solo nel 3% dei pazienti. Sulla base
della nostra scoperta, abbiamo ipotizzato che interno ischemia dell'arteria
uditiva provoca raramente coinvolgimento selettivo delle anteriore vestibolare
un'arteria o arteria cocleare. A differenza di una disfunzione
dell'orecchio interno di una causa virale, che può comunemente presenti come
vestibolare isolato (cioè, neurite vestibolare) o perdita cocleare (vale a
dire, sordità improvvisa), una disfunzione labirintica di una causa vascolare
raramente si traduce in perdita isolata di vestibolare o della funzione uditiva. Così,
quando improvvisa insorgenza di isolati prolungato vertigini o dell'udito
perdita si è verificata in pazienti con fattori di rischio vascolare,
compromissione vascolare all'orecchio interno è meno probabile considerata. Tuttavia,
quando la perdita audiovestibular combinato verificato in pazienti con
vertigini prolungata, la causa vascolare era altamente sospetto. La nostra
scoperta ha sostenuto l'ipotesi che improvvisa insorgenza di isolati sindrome
audiovestibular con vertigini e perdita dell'udito può derivare da un evento
vascolare (cioè infarto labirintico). Infatti, AICA territorio infarto può
produrre un modello unico di perdita vestibuloauditory in quanto la perdita
combinato di entrambi funzione vestibolare e coclea, piuttosto che vestibolare
isolato o perdita cocleare, sarebbero più comunemente prevedibile se arteria
uditivo interno è coinvolto.
Sebbene CP
unilaterale indica generalmente una lesione nelle strutture vestibolari
periferici dal labirinto omolaterale al nervo vestibolare, compresa la zona di
ingresso principale all'incrocio pontomedullary, è ben noto che la zona di
ingresso principale dell'ottavo nervo cranico ha una fitta rete di anastomotica
vasi derivanti dalla arteria laterale midollare, AICA, e inferiore pontina
arteria laterale. 21,22 Così, isolato infarto
focale in quella zona è altamente improbabile. Inoltre, per quanto a
nostra conoscenza, non vi era alcuna relazione di una sindrome vertigini
vascolare a causa di infarto focale nella zona di ingresso della radice del
nervo vestibolare.Infatti, riteniamo che la possibilità di CP associata con una
lesione nella zona voce principale dell'ottavo nervo cranico era estremamente
basso nei nostri pazienti con infarto AICA.
Il nostro studio ha
diversi limiti. Abbiamo solo la funzione del labirinto vestibolare
superiore con orizzontale canale semicircolare valutato utilizzando test
calorico e non tentare di valutare la funzione del labirinto vestibolare
inferiore che comprende il canale semicircolare posteriore e sacculo con le
loro fibre afferenti. In secondo luogo, perché il nostro studio ha incluso
solo i pazienti con documentata AICA territorio infarto alla risonanza
magnetica, lo spettro della disfunzione audiovestibular resta da chiarire in
isolato infarto labirintica.
In terzo luogo,
perché alcuni dei nostri pazienti ha avuto anche una lesione nei settori
diversi da territorio AICA, alcuni dei segni neurologici, tra cui anomalie
oculari motori, arti atassia, e gravi disturbi andatura con la caduta, possono
essere ricondotti ad una disfunzione delle aree forniti da le arterie diverse
da AICA. Infine, perché MRA nella maggior parte dei pazienti non può
adeguatamente visualizza posteriore inferiore arteria cerebellare / AICA, e
loro rami più piccoli, ulteriori studi che utilizzano l'angiografia
convenzionale sono necessari per valutare lo stato vascolare del AICA e
posteriore inferiore arteria cerebellare in AICA infarto.
.In
conclusione, infarto nel territorio AICA può presentare con un ampio spettro di
disfunzioni audiovestibular Considerando la bassa incidenza di cocleare
selettiva o coinvolgimento vestibolare in AICA infarto, compromissione
vascolare sembra dare luogo a perdita combinata di funzioni uditive e
vestibolari, mentre malattia virale comunemente presenta come vestibolare
isolato (cioè, neurite vestibolare) o perdita cocleare (vale a dire, sordità
improvvisa).
Lee H, Sohn SI, Jung DK, Cho YW, Lim JG, Yi SD, Lee SR, Sohn CH,
Baloh RW. Sudden deafness and anterior inferior cerebellar artery
infarction. Stroke.2002; 33: 2807–2812.
Roquer J, Lorenzo RJ, Pou A. The
anterior inferior cerebellar artery infarcts: a clinical-magnetic resonance
imaging study. Acta Neurol Scand. 1998; 97:225–230.
Lee H, Whitman GT, Lim JG, Lee SD,
Park YC. Bilateral sudden deafness as a prodrome of anterior inferior
cerebellar artery infarction. Arch Neurol. 2001;58: 1287–1289.
Lee H, Cho YW. Auditory disturbance as
a prodrome of anterior inferior cerebellar artery infarction. J
Neurol Neurosurg Psychiatry. 2003; 74: 1644–1648.
Lee H, Ahn BH, Baloh RW. Isolated
sudden deafness with vertigo as a sole manifestation of anterior inferior
cerebellar infarction. J Neurol Sci. 2004; 222:105–107.
Murakami T, Nakayasu H, Doi M, Fukada
Y, Hayashi M, Suzuki T, Takeuchi Y, Nakashima K. Anterior and posterior
inferior cerebellar artery infarction with sudden deafness and vertigo. J
Clin Neurosci. 2006; 13: 1051–1054.
Son EJ, Bang JH, Kang JG. Anterior
inferior cerebellar artery infarction presenting with sudden hearing loss and
vertigo. Laryngoscope. 2007; 117:556–558.
Ito H, Hibino M, Iino M, Matsuura K,
Kamei T. Unilateral hearing disturbance could be an isolated manifestation
prior to ipsilateral anterior inferior cerebellar artery infarction. Intern
Med. 2008; 47: 795–796.
Lee H, Kim HJ, Koo JW, Kim JS. Progression
of acute cochleovestibulopathy into anterior inferior cerebellar artery
infarction. J Neurol Sci. 2009; 278: 119–122.
Kim JS, Cho KH, Lee H. Isolated labyrinthine
infarction as a harbinger of anterior inferior cerebellar artery territory
infarction with normal diffusion-weighted brain MRI. J
Neurol Sci. 2009; 278: 82–84.
Matsushita K, Naritomi H, Kazui S,
Watanable Y, Okazaki H, Kuriyama Y, Sawada T. Infarction in the anterior
inferior cerebellar artery territory: magnetic resonance imaging and auditory
brain stem responses. Cerebrovasc Dis. 1993; 3: 206–212.
 Malattia
dei piccoli vasi:
Malattia
dei piccoli vasi: