Sindrome di Usher La sindrome
Graefe-Usher La sindrome sordità-retinite pigmentosa La sindrome Hallgren
Che cos'è la sindrome di Usher?
Storia.
Quanto è diffusa la sindrome di Usher?
Che cosa provoca la sindrome di Usher?
Quali
sono le caratteristiche dei tre tipi di sindrome di Usher?
Quali geni sono legati alla sindrome di
Usher?
Come viene diagnosticata la sindrome di Usher?
Come le persone ereditano la sindrome di Usher?
È disponibile un test genetico per
la sindrome di Usher?
Che trattamento è disponibile per
la sindrome di Usher?
Implicazioni psicosociali della sindrome di Usher.
Il
ruolo dell’ audiologo.
Altre forme di ipoacusia associate a perdita della vista.
Casi individuali .
Bibliografia.
Che cos'è la sindrome di Usher? Fig.1
- trasmissione autosomica recessiva
- sordità neurosensoriale congenita (raramente progressiva)
- retinite pigmentosa (RP)
- eterogeneità genetica - Tipi I, Il, III
|
Sindrome di Usher
|
Modalità di trasmissione
|
Tipo di deficit uditivo
|
Anomalie associate
|
|
⋅⋅⋅ 3-5% delle
sordità
⋅⋅⋅ 18% delle
retiniti
pigmentose
⋅⋅⋅ 50% dei pz
sordo-ciechi
|
A.
R.
Eterogeneità
genetica
(fenomeno per cui mutazioni
in loci genetici diversi possono avere lo stesso effetto fenotipico) 12 loci
genici e 9 geni
identificati come
causa
di S. di Usher
|
|
Tipo
1 ipoacusia
neurosensoriale
congenita profonda
|
|
Tipo
2 (>50%) ipoacusia
congenita
neurosensoriale
da
moderata
a profonda, in caduta sulle frequenze acute
|
|
Tipo
3 (rara) ipoacusia
neurosensoriale
che
evolve
nel tempo fino a profonda
|
|
|
1
Retinite pigmentosa ad insorgenza
nella prima decade
Areflessia
vestibolare
Ritardo motorio
|
|
2Retinite pigmentosa ad insorgenza
nella
seconda decade
Normale funzione vestibolare
|
|
3Retinite pigmentosa che si
manifesta
solitamente alla pubertà
Funzione
vestibolare variabilmente
affetta
|
|
La sindrome di Usher è una condizione genetica che comporta la perdita dell'udito neurosensoriale
e retinite pigmentosa (RP) ,è il tipo più comune di perdita di udito autosomica
recessiva sindromica. Sebbene sia considerata
una malattia rara, è la causa più frequente di sordo-cecità negli esseri
umani. Una sindrome è una malattia o un disturbo che ha più
di una caratteristica o sintomo. Quando la
perdita dell'udito è accompagnata da altri riscontri clinici, è spesso
classificato come una sindrome, e ci sono più di 400 tali sindromi descritte da
Gorlin, Torell, e Cohen (1995). la sindrome di Usher è una
condizione caratterizzata da perdita progressiva dell'udito o sordità e perdita
della vista da retinite pigmentosa (RP). Esistono molte forme di s di
Usher, ma tre casi su quattro rientrano nel I tipo Questo si presenta con una
sordità congenita profonda, areflessia vestibolare bilaterale responsabile di
un ritardo motorio di deambulazione (attorno a 18-24 mesi), e retinite che si
manifesto più tardivamente, ma sempre nell’infanzia, I primi sintomi visivi
consistono in difficoltà visive in penombra, spesso evidenti solo nella seconda
decade d’età, ma un esame del fondo oculare ed un elettroretinogramma, possono
essere diagnostici anche in età infantile Questa forma richiede una diagnosi
precoce, entro i 2-3 anni, perché un impianto cocleare può ridurre di molto le
difficoltà comunicative dovute al sommarsi di difetto uditivo e visivo .Nella s
di Usher tipo Il la sordità è meno grave, stabile, prevalentemente sulle
frequenze acute Anche la retinite interviene più tardivamente ed i difetti
vestibolari sono assenti .Nella s di Usher tipo III la sordità è progressiva,
mentre i sintomi vestibolari e la retinite pigmentosa insorgono in età
variabili .Sono stati localizzarti 11 geni diversi responsabili di queste
forme, dei quali otto identificati: 5 per il tipo I, due per il tipo Il, uno
per il tipo III. La maggior parte delle s. di Usher sono causate da mutazioni
dei geni MYO7A, CDH23, USH2A che codificono per proteine importanti per la
costituzione delle cellule cigliate e della matrice extracellulare della
coclea. La condizione prende il nome da un oculista britannico, CH Usher, che
in un articolo nel 1914 descrisse diversi casi in cui si è sottolineato il
legame tra sordità congenita e RP. Tuttavia, nel lontano 1860 lavoratori,
come von Graef Lo screening uditivo neonatale ha ridotto l'età di
identificazione dei bambini con perdita uditiva 12-18 mesi a 6 mesi o meno
(Harrison & Rousch, 1996), ma la diagnosi di sindrome di Usher, con la sua
devastante perdita della vista, tipicamente ritardi 5-10 anni dietro l'identificazione
della perdita dell'udito (Kimberling & Lindenmuth, 2007). Così, mentre
i genitori imparano di perdita uditiva del loro bambino relativamente presto,
senza diagnosi differenziale si procede con le decisioni critiche relative
all'intervento, comunicazione e opzioni educative ignari che il loro bambino
finirà per essere cieco. A causa dello screening uditivo neonatale,
audiologi sono spesso contatto primario della famiglia. Pertanto,
audiologi sono in grado di migliorare i risultati di diagnosi differenziali per
i bambini con sindrome di Usher. Questo articolo è rivolto ad accrescere
la comprensione audiologi 'della presentazione audiologico e visivo, criteri
diagnostici e strategie di intervento coinvolti con la sindrome di Usher.
Storia
La sindrome di Usher è chiamato dopo l'oculista scozzese Charles
Usher , che ha esaminato la patologia e
la trasmissione di questa malattia nel
1914 sulla base di 69 casi. Tuttavia, è stato descritto nel 1858
da Albrecht von Gräfe , (un pioniere della
moderna oftalmologia) e Liebreich a Berlino erano a
conoscenza del legame tra sordità congenita e RP, soprattutto nei matrimoni
consanguinei.. Egli ha riferito il caso di un paziente sordo
con retinite pigmentosa , che aveva due
fratelli con gli stessi sintomi. Tre anni più tardi(1861), uno dei suoi
studenti, Richard Liebreich , ha esaminato la
popolazione di Berlino che avevano sordità con retinite
pigmentosa. Liebreich ha notato che la sindrome di Usher era di tipo
recessivo, dal momento che i casi di combinazioni cieco sordità si sono
verificati in particolare nei fratelli dei matrimoni consanguinei o in famiglie
con pazienti in diverse generazioni. Le sue osservazioni hanno fornito le
prime prove per la trasmissione accoppiata di cecità e sordità, dal momento che
non potrebbe essere trovati casi isolati delle due patologie negli alberi
genealogici.
La perdita della vista è causata da una malattia dell'occhio chiamata
retinite pigmentosa (RP), che colpisce lo strato di tessuto sensibile alla luce
nella parte posteriore dell'occhio (retina). Perdita della vista si
verifica in quanto le cellule della retina che rilevano la luce si deteriorano
gradualmente. Di solito, i bastoncelli della retina sono i primi ad essere
interessati, con conseguente precoce cecità notturna e la graduale perdita di
visione periferica. In altri casi, si verifica, la degenerazione precoce dei
coni della macula, portando ad una perdita di acuità centrale. In alcuni casi,
è risparmiata la visione foveale, portando ad una "visione a
ciambella"; visione centrale e periferica sono intatti, ma un anello esiste
intorno alla regione centrale in cui visione è compromessa . Inizia prima
la perdita della visione notturna, cecità notturna, questo disturbo può anche
essere accoppiato alla difficoltà ad adattarsi alla luce intensa od a rapidi
cambiamenti delle condizioni di luce, seguita da punti ciechi che si sviluppano
nella visione laterale (periferica). Nel corso del tempo, questi punti
ciechi si ingrandiscono e si fondono per produrre una visione a tunnel. In
alcuni casi di sindrome di Usher, la visione è ulteriormente compromessa dalla
opacità del cristallino dell'occhio (cataratta). Molte persone con
retinite pigmentosa mantiene una certa visione centrale per tutta la vita,
però.
Quanto è diffusa la sindrome di Usher?
Si pensa che la sindrome di Usher possa
essere responsabile dal 3 per cento, al 6 per cento di tutta la sordità
infantile e circa del 50 per cento dei casi di sordità-cecità negli
adulti. Si stima che la sindrome Usher di I tipo colpisce almeno 4 individui
ogni 100.000 persone. Negli Stati Uniti, i tipi
1 e 2 sono i tipi più comuni. Insieme, essi rappresentano circa il 90 al
95 per cento di tutti i casi di bambini che hanno la sindrome di Usher. Può
essere ancora più comune in alcune popolazioni etniche, come nelle etnie di
ascendenza ebraica Ashkenazi (dell'Europa centrale e orientale) e nella
popolazione Acadian in Louisiana. Si pensa che la sindrome di Usher di II Tipo
sia la forma più comune, anche se la frequenza di questo tipo è sconosciuto. Nella
maggior parte delle popolazioni la sindrome di Usher di Tipo III rappresenta
solo una piccola percentuale di tutti i casi di sindrome di Usher. Questa
forma della malattia ,tuttavia, è più comune nella popolazione finlandese, dove
rappresenta circa il 40 per cento di tutti i casi.
I ricercatori hanno
individuato tre principali tipi di sindrome di Usher, designati come tipo I,
II, e III. Questi tipi si distinguono per la loro gravità e l'età in cui i
segni ei sintomi compaiono. Il tipo I è ulteriormente suddivisa in sette
distinti sottotipi, indicati come tipi IA/IG. La sindrome di Usher di tipo
II ha almeno tre sottotipi descritti, designati come tipi IIA, IIB e IIC.
|

|
|
Fig.
1:Fotografia della retina di un paziente
con la sindrome di Usher (a sinistra) rispetto ad una retina normale (a
destra). Il nervo ottico (freccia) sembra molto chiaro, i vasi (stelle)
sono molto sottili e non vi è pigmento caratteristico, chiamato spicole ossee
(doppie frecce).
|
|
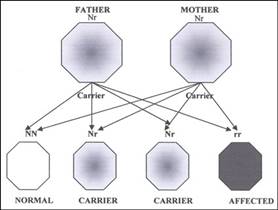 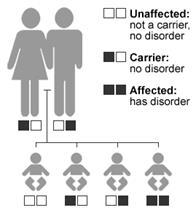 Fig.2a/b Fig.2a/b
|
|
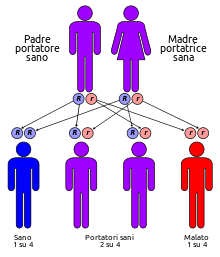
Le
malattie genetiche possono essere causate da un cambiamento (s) in un
gene. Ogni individuo ha due copie dello stesso gene. Malattie
genetiche vengono ereditate in modi diversi. La sindrome di Usher è una
malattia recessiva.Fig.2a/b
Mezzi Recessiva:
- una persona
deve ereditare un cambiamento nello stesso gene da ciascun genitore per
avere il disturbo
- una persona
con un gene mutato non ha il disturbo, ma può passare sia il mutato o il
gene invariata al suo bambino
Un
individuo con la sindrome di Usher solito:
- ha ereditato
un cambiamento nello stesso gene da ciascun genitore
Un
individuo che ha una cambiata Usher gene sindrome è chiamato un vettore.
Quando due portatori dello stesso gene sindrome di Usher hanno un figlio
insieme, con ogni nascita c'è un:
- 1-in-possibilità
su 4 di avere un bambino con la sindrome di Usher
- 2-in-possibilità
su 4 di avere un bambino che è un vettore
- 1-in-possibilità
su 4 di avere un bambino che non ha né la sindrome di Usher non è un
vettore
|
Che
cosa provoca la sindrome di Usher?
Sindrome di Usher è ereditata, il che significa
che si passa dai genitori ai figli attraverso i geni. Geni sono
localizzati in quasi ogni cellula del corpo. I geni contengono le
istruzioni che dicono le cellule cosa fare. Ogni persona eredita due copie
di ogni gene, uno da ciascun genitore. A volte i geni sono alterati o
mutati. Geni mutati possono indurre le cellule a agire in modo diverso
rispetto al previsto.
La sindrome di Usher è ereditata come carattere
autosomico recessivo. Il termine autosomica significa che il
gene mutato non è situato su uno dei cromosomi che determinano il sesso di una
persona; in altre parole, maschi e femmine possono avere la malattia e può
passarlo insieme ad un bambino. La parola recessiva significa che,
per avere la sindrome di Usher, una persona deve ricevere una forma mutata del
gene sindrome di Usher da ciascun genitore. Se un bambino ha una mutazione
in un gene (sindrome Usher), ma l'altro gene è normale, si prevede che avrà un
udito ed una visione normale '. Le persone con una mutazione in un gene
che può causare una malattia autosomica recessiva sono chiamati vettori, perché
"portano" il gene con una mutazione, ma non mostrano sintomi del
disturbo. Se entrambi i genitori sono portatori di un gene mutato per la
sindrome di Usher, avranno una probabilità su quattro di avere un figlio con la
sindrome di Usher con ogni nascita.
Di solito, i genitori che hanno un udito normale
e la visione non sanno se sono portatori di una mutazione del gene (sindrome di
Usher). Attualmente, non è possibile determinare se una persona che non ha una
storia familiare di sindrome di Usher è un vettore.
Quali
sono le caratteristiche dei tre tipi di sindrome di Usher?
Tipo 1
I bambini con diabete di tipo 1, e sindrome di Usher nascono
completamente sordi ( Fig. 3a) o perdono gran parte della loro udito
entro il primo anno di vita ed hanno gravi problemi di equilibrio. La perdita progressiva
della vista ,causata dalla retinite pigmentosa, si manifesta durante
l'infanzia. Questo tipo di sindrome di Usher comprende anche problemi dell'orecchio
interno che influenzano l'equilibrio. I
genitori dovrebbero consultare il proprio medico e altri professionisti della
salute dell'udito il più presto possibile per determinare il miglior metodo di
comunicazione per il loro bambino. L'intervento dovrebbe essere introdotto
presto, durante i primi anni di vita, in modo che il bambino può approfittare
della finestra unica di tempo durante il quale il cervello è più ricettivo per
l'apprendimento delle lingue, sia parlato o firmato. Se un bambino viene
diagnosticato con il tipo 1, sindrome di Usher presto, prima che lui o lei
perde la capacità di vedere, quel bambino ha maggiori probabilità di
beneficiare di tutta la gamma di strategie di intervento che possono aiutare
lui o lei di partecipare più pienamente nelle attività della vita.
La funzionalità vestibolare è caratterizzata da areflessia nella
sindrome di Usher Tipo I ,a causa dei problemi di equilibrio associati alla
sindrome di Usher di tipo 1, i bambini con questo disturbo di solito associato
a areflessia vestibolare e ritardo nelle tappe dello sviluppo, come nel
controllo del capo e nell'acquisizione della stazione seduta e della deambulazione
autonoma, sono lenti a sedersi senza supporto e di solito non camminano
autonomamente prima dei 18 mesi di età(Moller, Kimberling, Davenport, Priluck,
& White, 1989). Questi bambini di solito iniziano a sviluppare
problemi di visione nella prima infanzia, quasi sempre dal momento in cui
raggiungono 10 anni. Questa forma richiede una diagnosi precoce, entro i 2-3
anni, perché un impianto cocleare può ridurre di molto le difficoltà
comunicative dovute al sommarsi di difetto uditivo e visivo
Problemi di visione più spesso iniziano con difficoltà a vedere di
notte, Cecità notturna è tipicamente appare a 10 anni, quindi questi bambini
possono avere paura del buio e sono spesso descritti come goffi perché urtano,
o inciampare. Il deterioramento significativo del campo visivo e l'acutezza
inizia tra la seconda e la terza decade di vita, con la cataratta essendo una
complicanza comune (Edwards, Fishman, Anderson, Boschetto, e Derlackie, 1998;
Piazza, Fishman, Sugata, Derlacki, e Anderson, 1986; A. Sadeghi, Eriksson,
Kimberling, Sjostrom, e Moller, 2006) ma tendono a progredire rapidamente fino
a quando la persona è completamente cieco.
|

|
|
Fig. 2a. Tipici risultati audiometrici nella sindrome di
tipo 1 Usher.
|
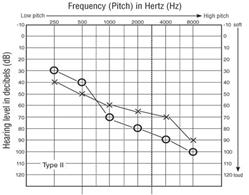
Tipo 2
La sindrome di Usher di tipo 2 (circa il 60% dei casi), è
caratterizzata da perdita dell'udito dalla nascita Fig.2b ,(la sordità è
meno grave, stabile, prevalentemente sulle frequenze acute) e progressiva
perdita della vista che inizia in adolescenza o in età adulta e solitamente il
quadro è meno grave rispetto al 1 tipo, non associata a alterazioni
vestibolari,. La perdita di udito associata a questa forma di sindrome di
Usher varia da una forma lieve ad una grave e colpisce soprattutto i toni
alti. I bambini affetti hanno problemi di udito, suoni del linguaggio
morbido elevate, come quelle delle lettere S e t. Il grado di perdita
dell'udito varia all'interno e tra le famiglie con questa condizione. A
differenza di altre forme di sindrome di Usher, le persone con diabete di tipo
II non hanno problemi di equilibrio causata da problemi dell'orecchio interno.
|
|
|
Figura 2b. Tipici risultati audiometrici di tipo II
sindrome di Usher.
|
Tipo 3
I bambini con sindrome di Usher tipo 3 (<3% dei casi, diffusa
soprattutto nelle popolazioni Finlandesi e negli Ebrei Ashkenaziti), hanno un
udito normale alla nascita, che progredisce durante l'adolescenza ,dopo lo
sviluppo del linguaggio, richiedono apparecchi acustici con l'età adulta, la
sordità è progressiva, l’ipoacusia è simile a quella descritta per il tipo II
,ma compare attorno ai 3-5 anni e presenta una progressività nel corso degli
anni (Kumar et al.. 1984).la perdita dell'udito e perdita della vista sono
progressivi a partire dai primi decenni di vita a differenza delle altre forme
di sindrome di Usher. Sebbene la maggior parte dei bambini con il disturbo
hanno un equilibrio normale o quasi normale, alcuni possono sviluppare problemi
di equilibrio in seguito, la disfunzione vestibolare si ha nella metà dei casi,
i pazienti presentano un quadro variabile caratterizzato da progressivo
peggioramento (Fishman. 1979: Fishman et al.. 1983). Con la mezza età, gli
individui più colpiti sono sordi. Anche la perdita della vista causata dalla
retinite pigmentosa si sviluppa nella tarda infanzia o nell'adolescenza. Le
persone con sindrome di Usher di tipo III possono anche avere difficoltà con
l'equilibrio a causa di problemi dell'orecchio interno. Questi problemi
variano tra gli individui affetti, Nella s di Usher tipo III i bambini hanno
un udito normale alla nascita, mentre i sintomi vestibolari e la retinite
pigmentosa insorgono in età variabili
|
Tipo 1
|
Tipo 2
|
Tipo 3
|
|
Udito
|
Sordità profonda in entrambe
le orecchie dalla nascita
|
Da moderata a grave perdita
di udito dalla nascita
|
Normali alla
nascita;progressiva perdita durante l'infanzia o adolescenza
|
|
Visione
|
Diminuzione della visione
notturna prima dei 10 anni
|
Visione notturna Diminuzione
inizia nella tarda infanzia o adolescenza
|
Varia in gravità; problemi
di visione notturna spesso iniziano negli anni dell'adolescenza
|
|
Vestibolare
funzione )
|
Problemi di equilibrio dalla
nascita
|
Normale
|
Normale al caso quasi normale,
di successivi problemi
|
|
Quali geni sono legati alla sindrome di
Usher?
Mutazioni nel CDH23, CLRN1, GPR98,
MYO7A, PCDH15, USH1C, USH1G, ei geni USH2A causano la
sindrome di Usher.
I geni legati alla sindrome di Usher
forniscono istruzioni per fare proteine che svolgono ruoli
importanti nella udito normale, equilibrio, e la visione. Funzionano nello
sviluppo e nel mantenimento delle cellule ciliate, che sono cellule sensoriali
dell'orecchio interno che aiutano a trasmettere segnali di movimento del suono
e al cervello. Nella retina, questi geni sono coinvolti nel determinare la
struttura e la funzione delle cellule sensibili alla luce bastoncelli e
coni. In alcuni casi, l'esatto ruolo di questi geni in ascolto e visione è
sconosciuta. La maggior parte delle mutazioni responsabili della sindrome
Usher portare ad una perdita di cellule ciliate nell'orecchio interno e una
graduale perdita di coni e bastoncelli della retina. La degenerazione di
queste cellule sensoriali provoca perdita di udito, problemi di equilibrio, e
la perdita di visione caratteristica di questa condizione.
Usher sindrome tipo che posso derivare da
mutazioni nel CDH23, MYO7A, PCDH15, USH1C, o geneUSH1G. Almeno
altri due geni identificati anche causare questa forma di sindrome di Usher.
La sindrome di Usher di tipo II è causata da
mutazioni in almeno quattro geni. Solo due di questi geni,USH2A e GPR98 (chiamato
anche VLGR1), sono stati identificati.
Le mutazioni in almeno due geni sono
responsabili della sindrome di Usher tipo III; tuttavia, CLRN1 è
l'unico gene che è stato identificato.
Per saperne di più sul CDH23 , CLRN1 , GPR98 , MYO7A , PCDH15 , USH1C , USH1G , e USH2Ageni.
Nella sindrome di Usher tipo I sono stati
identificati 7 sedi di linkage e 4 geni (MYO7A. USH1C. CDH23. PCDH15) a carico
dei quali sono state osservate varie mutazioni, con sordità congenita profonda,
areflessia vestibolare bilaterale responsabile di un ritardo della
deambulazione (deambulazione dopo 18 mesi) e retinite che si sviluppa durante
l'infanzia. I primi segni visivi sono dei disturbi della visione in penombra,
spesso verso l'inizio del secondo decennio di vita, ma l'esame sistematico del
fondo dell'occhio può consentire la diagnosi molto prima di questa età, fin dai
3-4 anni. L'esame più precoce è l'elettroretinogramma, patologico prima dei
primi segni nel fondo dell'occhio. La sindrome di Usher di tipo I è
un'indicazione all'impianto cocleare precoce per ottenere una comprensione del
linguaggio senza lettura labiale in questi bambini che, in età adulta, avranno
una rilevante compromissione visiva. Porre la diagnosi attraverso l'esame del
fondo dell'occhio a 4 anni è quindi già una situazione tardiva. In linea di
principio l'esame oftalmologico esteso al fondo dell'occhio deve essere
sistematico e ripetuto nel bambino e nell'adulto sordi, e ogni sordità profonda
congenita con ritardo della deambulazione senza eziologia evidente deve fare
eseguire un elettroretinogramma, anche se il fondo dell'occhio è normale La
funzionalità vestibolare è caratterizzata da areflessia nella sindrome di Usher
Tipo I ,da normoreflessia nella sindrome di Usher Tipo II e da risposte che
vanno progressivamente peggiorando nella sindrome di Usher Tipo III (Kumar et
al. 1984: Möller et al.. 1989)
Nella sindrome di Usher tipo II sono state
osservate varie mutazioni a carico dii gene (USH2A); tuttavia è verosimile che
vi sia una eterogeneità genetica dal momento che sono state identificate 2
ulteriori sedi di linkage, la sordità è in media grave, non progressiva,
predominante sulle frequenze acute, la retinite un poco più tardiva e i segni
vestibolari assenti. Nella Usher tipo III è stato recentemente individuato 1
gene (USH3) a carico del quale sono state osservate varie mutazioni (Tab. III).
la sordità è progressiva, i segni vestibolari e l'età di inizio della retinite
sono variabili (per una rassegna, vedi [Grøndahl J (1987). 8]).
A tutt'oggi sono stati localizzati 12 geni diversi responsabili di
queste forme Geni e le proteine che codificano sono stati identificati per 7
dei 12 loci., cinque per la sindrome di Usher tipo I, due per il tipo II e uno
per il tipo III (Tabella II/III).
MYO7A e CDH23 rappresentano rispettivamente il 30 e 29% dei casi
di Usher tipo I e USH2A il 40% dei tipi II(Ouyang X.) La diagnosi
molecolare non è eseguita di routine e la diagnosi è essenzialmente clinica.
I geni che causano la sindrome di Usher sono
MY07A, USH1C, CDH23, PCDH15,e SANS, che causa USH1, USH2A (che causa USH2 ), e
USH3A (che provoca USH3 ). Una mutazione, denominata R245X, del PCDH15 gene può
rappresentare una grande percentuale di USH1 casi in Ashkenazi popolazione
ebraica di oggi La maggior parte delle s. di Usher sono causate da mutazioni
dei geni MYO7A, CDH23, USH2A che codificano per proteine importanti per la
costituzione delle cellule cigliate e della matrice extracellulare della
coclea.
Tab,
II. Genetica della s, di Usher.
|
Locus
|
Localizzazione
|
Gene
|
Marker
di screening
|
Most
lmportant Reference
|
OMIM
Entry
|
|
USH1A
|
14q32
|
Unknown
|
D14S250,
D14S260, D14S292, D14S78
|
Kaplan et al., 1992
|
276900
|
|
USH1B
|
11q135
|
MYO7A
|
Di 1S906, Dl 1S911, D11S52, OMP-CA
|
Weil et al,1995
|
276903
|
|
USH1C
|
hp15.1
|
USH1C
|
D11S902,D11S921, D11S899,D11S861
|
Smith et
al,, 1992
Verpyetal,2000
Bitner-Glindzicz et aL,
2000
|
276904
|
|
USH1D
|
10q
|
CDH23
|
D10S529,
D10S202, D10S573
|
Wayne et
al,, 1996
Bork et al., 2001
Bolz et al,, 2001
|
601067
|
|
USH1E
|
21q
|
Unknown
|
D21S1884,
D21S1257,
D21 2S265,
D21S1258
|
Chaib et
al, 1997
|
602097
|
|
USH1F
|
10q2122
|
PCDH15
|
D10S199,
D10S578, D10S596
|
Ahmed et
aL, 2001
Alagramam et al.,,
2001
|
602083
|
|
USH1G
|
17q24-25
|
Unknown
|
|
Mustapha
et al.., 2002
|
|
|
USH2A
|
1q41
|
USH2A
|
D1S229,
D1S490, D1S237, D1S474
|
Kimberling et al,
1990
Eudy et al., 1998
|
276901
|
|
USH2B
|
3p2324..2,
|
Unknown
|
D3S1578, D3S3647, D3S3658
|
Hmani et al,, 1999
|
276905
|
|
USH2C
|
5q14.3-q21.3
|
Unknown
|
D5S428, D5S421
|
Pieke-Dahl et al., 2000
|
605472
|
|
USH 3
|
3q21-q25
|
USH3
|
D3S1299, D3S1555, D3S1280, D3S1 279
|
Sankila et
al, 1995 Joensuu et al., 2001
|
276902
606397
|
Geni associati con la sindrome
di Usher Tab, III.
Come viene diagnosticata la sindrome di Usher?
Poiché la sindrome di Usher colpisce l'udito, equilibrio, e la
visione, la diagnosi del disturbo di solito include la valutazione di tutti e
tre i sensi. Misure comportamentali e oggettive del sistema uditivo sono tecniche
familiari per l'audiologo. Proprio come con le misure acustiche, la
valutazione della funzione visiva può essere comportamentale (cioè, l'acuità e
campo visivo) o oggettiva. Una videoelectronistagmografia (VNG) che misura
i movimenti involontari degli occhi potrebbe evidenziare dei problemi di
equilibrio. I tests obiettivi comprende l'esame diretto della retina, dove un
oculista troverà vasi sanguigni scoloriti, un pallore cereo, e grumi di cellule
retiniche morte chiamati spicole ossee (Carr & Noble, 1981). Tuttavia,
questi risultati fisici non sono evidenti fino a ben dopo che il paziente è
sintomatico. La prova definitiva di RP è un elettroretinogramma
(ERG). Come una risposta uditivi del tronco encefalico, l'ERG è una
risposta evocata ma dai coni e bastoncelli della retina. Poiché il test
richiede l'inserimento di una lente a contatto matrice / elettrodo, anestesia
generale è necessaria per i bambini, mentre farmaci topici possono essere utilizzati
con gli adulti. Anche se un bambino può avere una visione ancora
relativamente buona, l'ERG sarà ridotto o assente quando sindrome RP / Usher è
presente. Vi è qualche evidenza che mentre la perdita di ampiezza del ERG
è simile tra Usher tipo I e tipo II, il ritardo implicito si può distinguere
tra i tipi (Seeliger, Zrenner, Apfelstedt-Sylla, e Jaissle, 2001). Un ERG
presenta un vantaggio diagnostico distinto che sarà anormale molto prima dei segni
fisici di morte cellulare (spicole ossee) che compaiono sulla
retina. Questo spiega perché un oculista spesso non fa la diagnosi con un
giovane paziente, in quanto il bambino si esibirà bene nel test dell'acuità
visiva e le sue retine sotto esame diretto appariranno normali.
Valutazione degli occhi può includere un test del campo visivo per
misurare la visione di una persona periferica, un elettroretinogramma (ERG) per
misurare la risposta elettrica delle cellule fotosensibili dell'occhio, e un
esame della retina per osservare la retina e altre strutture nella parte
posteriore l'occhio. La diagnosi precoce della sindrome di Usher è molto
importante. Prima che i genitori sanno che il loro bambino ha la
sindrome di Usher, prima il bambino potrà iniziare programmi di formazione
educativa speciali per gestire la perdita di udito e della vista.
Come le persone ereditano la sindrome di Usher?
APPROFONDIMENTO
Questa condizione è ereditata come
carattere autosomico recessivo, il che significa che entrambe le copie del gene
in ogni cellula hanno mutazioni. I genitori di una persona con una
malattia autosomica recessiva portano ciascuno una copia del gene mutato, ma in
genere non mostrano segni e sintomi della malattia.
Dove posso trovare informazioni circa la
diagnosi o la gestione della sindrome di Usher?
Tali risorse riguardano la diagnosi o il
trattamento della sindrome di Usher e possono includere fornitori di
trattamento.
- Gene Recensione: Sindrome di Usher di tipo 1

- Gene Recensione: Sindrome di Usher di tipo 1
- Gene Recensione: Sindrome di Usher di tipo 2
- Gene Recensione: Sindrome di Usher di tipo 2
- Genetica Registro Test: Retinite
pigmentosa Sindrome-sordità
- Genetica Registro Test: La sindrome di Usher,
tipo 1
- Genetica Registro Test: La sindrome di Usher,
tipo 1C
- Genetica Registro Test: sindrome di Usher,
tipo 1D
- Genetica Registro Test: La sindrome di Usher,
tipo 1E
- Genetica Registro Test: La sindrome di Usher,
tipo 1F
- Genetica Registro Test: La sindrome di Usher,
tipo 1G
- Genetica Registro Test: La sindrome di Usher,
tipo 2A
- Genetica Registro Test: La sindrome di Usher,
tipo 2C
- Genetica Registro Test: La sindrome di Usher,
tipo 3
- Genetica Registro Test: Usher sindrome tipo ID /
F, CDH23/PCDH15, digenica
- MedlinePlus Enciclopedia:
Retinite pigmentosa
È disponibile un test genetico per la sindrome di Usher?
Finora, sono stati trovati 11 loci genetici
(un segmento del cromosoma in cui si trova un certo gene) per causare la
sindrome Usher, e sono stati individuati nove geni che causano la
malattia. Sono:
- Tipo 1 La sindrome di
Usher: MY07A, USH1C, CDH23, PCDH15, SANS
- Tipo 2 La sindrome di
Usher: USH2A, VLGR1, whrn
- Tipo 3 sindrome di
Usher: USH3A
Con così tanti possibili geni coinvolti nella sindrome di Usher,i test
genetici per la malattia non possono sono effettuati in modo
capillare. La diagnosi di sindrome di Usher è di solito effettuata
attraverso i tests dell'udito, dell'equilibrio, e della visione. Il test
genetico per alcuni dei geni identificati è clinicamente disponibile. Per
conoscere i laboratori che effettuano test clinici, visitare il sito Web www.GeneTests.org e
cercare la directory laboratorio digitando il termine "sindrome di
Usher". Il test genetico per ulteriori geni sindrome di Usher
potrebbe essere disponibile attraverso studi di ricerca clinica. Per
informazioni sulle sperimentazioni cliniche che includono il test genetico per
la sindrome di Usher, visitare il sito Web www.clinicaltrials.gov e
digitare il termine di ricerca "sindrome di Usher" o "test
genetici Usher".
Condotta da tenere
L'esame oftalmologico esteso al fondo dell'occhio deve
essere sistematico e ripetuto nel bambino e nell'adulto sordi e ogni sordità
profonda congenita con ritardo della deambulazione senza eziologia evidente
deve fare eseguire un elettroretinogramma, anche se il fondo dell'occhio è
normale. La diagnosi precoce è molto importante al fine del programma
terapeutico-riabilitativo. La prognosi infatti è caratterizzata dalla comparsa
di una cecità attorno ai 3 0-40 anni di età che andrà a sommarsi al deficit
uditivo. compromettendo notevolmente il grado di autonomia del soggetto
Che trattamento è disponibile per la sindrome
di Usher?
Attualmente non esiste un trattamento medico per prevenire, rallentare la
progressione della, o inibire la trasmissione di sindrome di
Usher. Impianto cocleare è una valida opzione provata per le persone con
sindrome di Usher. Gli individui con tipo I sono candidati dalla nascita,
mentre quelli con tipo II o III può diventare candidati nel corso del
tempo. Qualità significativo di miglioramento della vita sono state
dimostrate per i bambini con sordità congenita che ricevono impianti cocleari
(Schorr, Roth, e Fox, 2009), e l'impianto è stato dimostrato come benefico per
i bambini sordi con una varietà di disabilità associate (Berrettini et al.,
2008). Perché la perdita dell'udito nella sindrome di Usher è cocleare in
natura, l'impianto cocleare è una raccomandazione ragionevole e ha, di fatto,
dimostrato di beneficiare funzionamento uditiva e sociale dei bambini con
sindrome di Usher tipo I (Damon, Pennings, Snik, e Mylanus 2006 ; Liu et al,
2008)..
Ad
oggi non esiste alcun trattamento per la RP. Molti individui mantengono un
piccolo campo visivo utilizzabile anche in quinta o sesta decade di
vita. Vi sono prove preliminari che i fattori ambientali e dietetici
possono aumentare la longevità di visione utilizzabile. Questi includono
proteggere la retina dalla luce ultravioletta indossando 100% UVA / UVB
occhiali da sole e mantenere la salute globale ottimale. L'esercizio
fisico e la dieta, come ad esempio a base di pesce ricco di omega-3, sono stati
anche suggeriti (Berson, 2000). Gli studi sulla vitamina A e acido
docosaesaenoico (DHA) trattamenti hanno creato qualche polemica (Massof &
Finkelstein, 1993) e fino ad oggi sono stati solo condotti su persone con RP
non sindromica e alcuni Usher di tipo II pazienti (Berson et al sentite., 1993,
2004a , 2004b). I pazienti devono essere avvertiti concernente una
supplementazione di vitamina perché consumano troppo può causare ipervitaminosi
conseguente cecità, mancanza di crescita, e anche la morte. Le donne e le
donne incinte che possono diventare incinte non dovrebbero utilizzare
integratori come la vitamina A, perché possono causare difetti alla nascita nel
feto.
Implicazioni psicosociali della sindrome di
Usher
La
perdita sensoriale duale, in particolare la perdita progressiva della vista,
presenta molteplici sfide per le persone con sindrome di Usher e le loro
famiglie. In genere la perdita dell'udito è stata affrontata e le
decisioni sono state fatte sulla metodologia di comunicazione. Una cultura
identità Deaf può essere stata stabilita prima conoscenza della perdita della
vista imminente. I genitori sono spesso sopraffatti dalla seconda diagnosi
e credono che dovrebbero proteggere i loro figli da questa informazione fino a
tardi. Nel frattempo, il bambino può sospettare qualcosa ed avere ancora
più paura perché lui o lei non capisce. Condivisione di informazioni
adeguate all'età con i bambini è il miglior approcio. Non hanno bisogno di
sentirsi dire che stanno "diventando ciechi", ma hanno bisogno di
sapere che vedono in modo diverso e che ci sono strategie che li aiuteranno a
navigare nel loro mondo. Può essere giustificata l'aiuto di UNA consulenza
professionale. Vivere con la sindrome di Usher richiede adattamenti per tutta
la vita a cambiare lo stato di visione (Miner, 1995) e, in alcuni casi, cambiare
le capacità uditive. Come audiologi, non possiamo supporre ci sono
professionisti che hanno familiarità con l'impatto della perdita sensoriale
duale. La maggior parte degli specialisti della vista non conoscono la
perdita dell'udito e di fatto dipendono dall’audito per aiutare i loro pazienti
non vedenti. Gli individui con sindrome di Usher sono sempre sordi o con
problemi di udito prima che siano ipovedenti o non vedenti.
Il ruolo dell’ audiologo
La
diagnosi di una significativa perdita di udito in un bambino può così dominare
la nostra attenzione come audiologi che possiamo trascurare le altre condizioni
invalidanti. I genitori si affidano a audiologi per informazioni e
supporto nel lavoro con i loro bambini che hanno la perdita
dell'udito. Infatti, Steinberg e colleghi (2007) hanno identificato le
interazioni dei genitori con l'audiologo come tema importante nel loro esame
dei racconti dei genitori in materia di test genetici per la perdita dell'udito. Gli
autori hanno indicato che le aspettative dei genitori con gli audiologi
includono le informazioni riguardanti le risorse, sostegno e orientamento con i
rinvii e test, e permettendo ai genitori di adattarsi alle loro nuovi
ruoli. In sintesi, "audiologi sono spesso tenuti a consigliare,
guidare e aiutare i genitori attraverso le loro decisioni e le scelte"
(Steinberg et al., 2007, p. 64). Inoltre, l'American
Speech-Language-Hearing Association (ASHA) Linee guida per la
valutazione Audiologic dei bambini dalla nascita ai 5 anni di età consiglia
di audiologi dovrebbero ", come opportuno, discutere valutazioni speciali
aggiuntivi (ad esempio, la genetica, oculistica, sviluppo infantile) con i
genitori / tutori ei neonati primario fornitore di cure "(ASHA, 2004, p.
19). Ciò richiede che l'audiologo avere familiarità con l'epidemiologia
genetica della sordità e delle risorse di rinvio e di informazione.
Altre forme di ipoacusia associate a perdita della
vista
Secondo
la Gallaudet Research Institute (2008), il 40% di tutti i bambini con perdita
dell'udito neurosensoriale sono noti per avere una o più condizioni invalidanti
supplementari. In questo gruppo, il 10% ha problemi di vista non correggibile.
Differenziazione tra i bambini ipovedenti e coloro che sono sordo-ciechi è irto
di difficoltà dovute alla variabilità dell’udito residuo e della funzione visiva.
Perdita della vista, come la perdita dell'udito, varia notevolmente in gravità
e può essere misurata come acuità visiva da vicino e lontano, campo visivo
periferico, sensibilità al contrasto, visione dei colori, sensibilità
all'abbagliamento, e / o la visione notturna. Così come non si può veramente
prevedere la competenza comunicativa di un paziente da un audiogramma per i toni-
puri, un singolo parametro visivo non predice il funzionamento visivo.
Mentre Usher è la sindrome più frequentemente associato con RP, ci sono altre
condizioni che il clinico deve conoscere. La Sindrome di Alport si manifesta con
perdita dell’ udito, nefrite progressiva, e chiazze maculari che possono essere
fraintesi come RP, mentre la malattia di Refsum si presenta con anosmia,
sordità, e vero RP (Hamel, 2006). Altre condizioni come la sindrome di CHARGE,
citomegalovirus, toxoplasmosi, e la meningite può causare contemporaneamente
perdita dell’udito e della vista. 2008 Conte Child Nazionale dei bambini e dei
giovani che sono sordo-ciechi riporta 10.766 bambini di età compresa nascita
attraverso 21 anni sono stati identificati come (Consorzio Nazionale sulla
sordocecità, 2008) sordo-ciechi. Indipendentemente dalla eziologia, un
audiologo deve essere sensibile alla visione e le strategie suggerite qui per
lavorare con i pazienti con sindrome di Usher è applicabile a molti altri pazienti
con compromesse delle capacità visive.
Casi individuali da wikipendia
Casi individuali [ modifica ]
Una
donna di 31 anni, con la sindrome di Usher, Rebecca Alexander, è stato
profilato in Marie Claire nel novembre 2007. [21] Dopo
la laurea presso la University of Michigan con ottimi
voti, Alexander ha continuato alla Columbia University , dove ha conseguito
due gradi di master nella sanità pubblica e clinico sociale. Rebecca è un
membro attivo della sua comunità, collaborando con diverse associazioni di
beneficenza a New York. La dedizione di Rebecca come membro attivo della
sua comunità è stata in particolare riconosciuta quando è stata selezionata
come una "Comunità Hero" per correre con la torcia olimpica per i
Giochi di Atlanta olimpici del 1996 in onore del suo lavoro di volontariato per
il progetto Open Hand, un'organizzazione no-profit consegna pasti per le
persone che vivono con l'HIV / AIDS nella zona di San Francisco
Bay. Rebecca ha ricevuto la sua formazione psicoterapia psicodinamica attraverso
l'American Institute di Psicoanalisi. Attualmente lavora in uno studio
privato specializzato nel trattamento dei disturbi dell'umore e d'ansia,
disturbi alimentari, dipendenze, disabilità, e traumi. Mentre attualmente
facilita seminari di gruppo per la Foundation Fighting Blindness durante
le conferenze nazionali, Rebecca è anche in procinto di lanciare l'iniziativa
Usher III, un'organizzazione no profit dedicata alla scienza e alla ricerca che
cerca di trovare una cura per Usher III. Rebecca insegna ciclismo / spin
nelle classi interne con un forte seguito in alcune palestre di New York
City. È stata descritta sulla Today Show della NBC il 20 marzo 2009, che è
stato nominato per un premio Emmy nel settembre 2010. Lei è la sorella di NBC
News Nazionale Corrispondente Peter Alexander.
Christine "Coco" Roschaert [22] è
una persona conosciuta con la sindrome di Usher. Ha pubblicato un video
blog su YouTube, [23] e
recentemente è stato l'altoparlante kick-off per la Settimana della prevenzione
della sordità presso l' Università del Vermont . [24]Nel
2006, si è laureata con una laurea in Scienze della Comunicazione alla Gallaudet University ; lì, lei ha
fatto lo sciopero della fame come segno di protesta 2006 organizzata
dalla Gallaudet Stati Ora Movimento . [25] Roschaert
è ora in Nigeria per la fondazione del programma prima sordociechi .
Una
web-community, UsherLife ,
delle persone con sindrome di Usher è stata fondata il 1 ° febbraio 2005 da
Nick Sturley. Anche se centrata sulla Gran Bretagna , che offre risorse per tutte le
persone con sindrome di Usher. L'organizzazione sta ospitando regolarmente
ritrovi in Inghilterra, come il Usher Hood Pub a Nottingham [26] e
un viaggio a Brighton Pier. [27] Altre
persone con sindrome di Usher hanno inviato video sulla loro vita e le
condizioni su YouTube , in particolare . Ginny Paja-Nyholm[28] Nell'ottobre
2007, Candice, una mamma che vive in Texas, ha iniziato a bloggare sulle sue
due figlie, Jasmine e Rebecca;Rebecca che hanno la sindrome di Usher I. [29]
Catherine Fischer ha scritto un'autobiografia ben accolta di “crescere con la
sindrome di Usher in Louisiana”, intitolato Orchidea del Bayou. [30] Allo
stesso modo, Vendon Wright ha scritto due libri che descrivono la sua vita con
la sindrome di Usher, ero cieco, ma ora vedo [31] e attraverso
i miei occhi. [32] Louise
Boardman ha anche scritto un breve libro intitolato Mio figlio ha la
sindrome di Usher. [33]
Christian Markovic, un artista che vive con la sindrome di
Usher, gestisce una società, Fuzzy disegni wuzzy. [34]
Spencer Tracy figlio s 'Giovanni era una persona
ben conosciuta con la sindrome di Usher, che ha vissuto una vita piena. [35] IlJohn Tracy Clinic è stata fondata nel 1942
da sua madre Louise per offrire assistenza gratuita ai genitori dei bambini
udenti e bambini in età prescolare . [1]
Jacob Desormeaux, figlio del fantino di ippica Kent Desormeaux , ha la sindrome di
Usher. Jacob è nato sordo e sta progressivamente diventando
cieco. Kent ha dedicato la sua corsa nelle Belmont Stakes , che lui e il suo cavallo
darebbero Big Brown la Triple Crown, a suo figlio
Giacobbe. La famiglia ha avviato un'organizzazione per raccogliere fondi e
la consapevolezza della malattia. La sindrome di Usher è
sproporzionatamente comune tra i cajun della Louisiana a sud, come Desormeaux e Fischer, a
causa di una mutazione genetica tra i primi francesi Acadian coloni in Nova Scotia .
Elica del DNA co-scopritore e premio Nobel James D. Watson ha pubblicato[36] USH1B mutazioni
omozigoti, secondo il suo genoma. Non è chiaro il motivo per cui egli non
ha sviluppato la sindrome. Questa mancanza di penetranza genetica sostiene che
l'espressione del fenotipo della sindrome di Usher può essere più
complessa di quanto inizialmente ipotizzato.
Il
"Nalaga'at" israeliano (fare touch) sordo-ciechi Acting Ensemble è
composto da 11 attori sordo-cieco, molti dei quali sono diagnosticati con
sindrome di Usher. Il gruppo teatrale ha messo su diverse produzioni ed è
apparso sia localmente che in Israele e all'estero a Londra e Broadway. [37]
References
Alexander R, Grossman AJ (November 2007). "out of sight, out of
sound". Marie Claire 14 (11): 191–193.American Speech-Language-Hearing
Association. (2004). Guidelines for the audiologic assessment of
children from birth to 5 years of age. Available from www.asha.org/policy .
Bell J (1933). Retinitis Pigmentosa and Allied
Diseases (2nd ed.). London: Cambridge University Press.
Berrettini, S., Forli, F., Genovese, E., Santarello, R., Arslan,
E., Chilosi, A., & Cipriani, P. (2008). Cochlear
implantation in deaf children with associated disabilities: Challenges and
outcomes. International Journal of Audiology, 47, 199–208.
Berson, E. (2000). Nutrition and retinal
degeneration. International Ophthalmology Clinics , 40 (4),
93–111.
Berson, E., Rosner, B., Sandberg, M., Hayes, K.,
Nicholson, B., Weigel-DiFranco, C., & Willett, W. (1993). A randomized
trial of vitamin A and vitamin E supplementation for retinitis
pigmentosa. Archives of Ophthalmology, 111 , 761–772.
Berson, E., Rosner, B., Sandberg, M.,
Weigel-DiFranco, C., Moser, A., Brockhurst, R., et
al. (2004a). Clinical trial of docosahexaenoic acid in patients with
retinitis pigmentosa receiving vitamin A treatment. Archives of
Ophthalmology, 122 , 1297–1305.
Berson, E., Rosner, B., Sandberg, M.,
Weigel-DiFranco, C., Moser, A., Brockhurst, R., et
al. (2004b). Further evaluation of docosahexaenoic acid in patients
with retinitis pigmentosa receiving vitamin A treatment: Subgroup
analyses. Archives of Ophthalmology, 122, 1306–1314.
Boughman, J., Vernon, M., & Shaver, K. (1983). Usher
syndrome: Definition and estimate of prevalence from two high-risk
populations. Journal of Chronic Diseases, 36, 595–603.
Carr, R., & Noble, K. (1981). Retinitis pigmentosa. Ophthalmology,
88 (2), 169–172.
Chen, D. (2004). Young children who are
deaf-blind: Implications for professional in deaf and hard of hearing
services. Volta Review, 104 (4), 273–284.
Cohen, M., Bitner-Glindziez, M., & Luxon, L.
(2007). The changing face of Usher syndrome: Clinical implications. International
Journal of Audiology, 46, 82–93.
Damon, G., Pennings, R., Snik, A., &
Mylanus, E. (2006). Quality of life and cochlear implantation in Usher
syndrome type I. Laryngoscope, 116, 723–728.
Davenport S, Omenn G (1977). The
Heterogeneity of Usher Syndrome (volume 426 ed.). Amsterdam: Excerpta
Medica Foundation.
Edwards, A., Fishman, G., Anderson, R., Grove,
S., & Derlackie, D. (1998). Visual acuity and visual field impairment
in Usher syndrome. Archives of Ophthalmology, 116 , 165–168.
Fishman GA, Kumar A, Joseph ME, Torok N, and
Andersonj RJ (1983). "Usher's syndrome". Archives of Ophthalmology
101 (9): 1367–1374.
Gallaudet Research Institute. (2008). Regional and
national summary report of data from the 2007–08 annual survey of deaf and hard
of hearing children and youth. Washington, DC: Author.
Gerber, S; Bonneau, D;
Gilbert, B; Munnich, A; Dufier, JL; Rozet, JM; Kaplan, J (2006). "USH1A:
chronicle of a slow death". American Journal of Human Genetics 78 (2):
357–9.
Gorlin R, Tilsner T,
Feinstein S, Duvall AJ (1979). "Usher syndrome type III". Arch.
Otolaryngol. 105 (6): 353–354.
Grøndahl J (1987).
"Estimation of prognosis and prevalence of retinitis pigmentosa and Usher
syndrome in Norway". Clin. Genet. 31 (4):
255–264.
Gorlin, R., Torella, H., & Cohen, M.
(1995). Hereditary hearing loss and its syndromes. Oxford,
England: Oxford University Press
Hallgren B (1959).
"Retinitis pigmentosa combined with congenital deafness with
vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases:
Clinical and geneto-statistical survey". Acta Psychiatrica Scandinavica Suppl. 34 (138):
9–101.
Hamel, C. (2006). Retinitis pigmentosa. Orphanet Journal of Rare
Diseases, 1 :(40, 1 – 12).
Hammerschlag V (1907). "Zur Kenntnis der hereditaer-degenerativen
Taubstummen und ihre differential diagnostische Bedeutung". Z. Ohrenheilk.
54: 18–36.
Harrison, S., & Rousch, J. (1996). Age
of suspicion, identification, and intervention for infants and young children
with hearing loss: A national study. Ear and Hearing, 17, 55–62.
Hashimoto T, Gibbs D, Lillo C, Azarian SM,
Legacki E, Zhang XM, Yang XJ, Williams DS (2007). "Lentiviral gene
replacement therapy of retinas in a mouse model for Usher syndrome type
1B". Gene Therapy 14 (7): 584–594
Hicks, W., & Hicks, D. (1981). The
Usher's syndrome adolescent: Programming implications for school
administrators, teachers, and residential advisors. American Annals of
the Deaf, 26 , 422–431.
Hope CI, Bundey S, Proops D, Fielder AR (1997).
"Usher syndrome in the city of Birmingham — prevalence and clinical
classification". British Journal of Ophthalmology 81 (1): 46–53.
Innaccone, A., Kritchevsky, S., Ciccarelli, M., Tedesco, S.,
Macahuso, C., Kimberling, W., & Somes, G. (2004). Kinetics
of visual field loss in Usher syndrome type II. Investigative
Ophthalmology & Visual Science, 45 , 784–792.
Keats, B. (2002). Genes and syndromic
hearing loss. Journal of Communication Disorders, 33, 355–366.
Kimberling, W., & Lindenmuth, A. (2007). Genetics,
hereditary hearing loss, and ethics. Seminars in Hearing, 28 (3),
216–225.
Liebreich R (1861).
"Abkunft aus Ehen unter Blutsverwandten als Grund von Retinitis
pigmentosa". Dtsch. Klin. 13: 53.
Liu, X., Angeli, S., Rajput, K., Yan, D.,
Hodges, A., Eshraghi, A., et al. (2008). Cochlear implantation in
individuals with Usher type 1 syndrome. International Journal of
Pediatric Otorhinolaryngology, 72, 841–847.
Massof, R., & Finkelstein, D.
(1993). Supplemental vitamin A retards loss of ERG amplitude in retinitis
pigmentosa. Archives of Ophthalmology, 111, 751–754.
Merin S, Auerbach E (1976). "Retinitis
pigmentosa". Surv. Ophthalmol. 20 (5): 303–345.
doi:10.1016/S0039-6257(96)90001-6
Mets MB, Young NM, Pass A, Lasky JB (2000)."Early
diagnosis of Usher syndrome in children". Transactions of the American
Ophthalmological Society 98: 237–45.
Miner, I. (1995). Psychosocial implications
of Usher syndrome, type I, throughout the life cycle. Journal of Visual
Impairment & Blindness, 89 (3), 287–296.
Moller, C., Kimberling, W., Davenport, S.,
Priluck, L., & White, V. (1989). Usher syndrome: An otoneurologic
study. Laryngoscope, 99, 73–79.
National Consortium on Deaf-Blindness. (2008). National
Child Count of Children and Youth Who Are Deaf-Blind . Retrieved
December 2, 2009, from www.nationaldb.org/documents/products/2008-Census-Tables.pdf [PDF].
Otterstedde CR, Spandau U,
Blankenagel A, Kimberling WJ, Reisser C (2001). "A new clinical classification
for Usher's syndrome based on a new subtype of Usher's syndrome type
I". Laryngoscope 111 (1): 84–86.
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson
SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton V, Brown SD, Balkany T, Liu XZ
(2005). "Characterization of Usher syndrome type I gene mutations in an
Usher syndrome patient population". Hum Genet 116 (4): 292–299.
Pakarinen L, Tuppurainen K, Laipapala P,
Mäntyjärvi M, Puhakka H (1996). "The ophthalmological course of Usher
syndrome type III". International Ophthalmology 19 (5): 307–311.
Petit, C (2001). "Usher syndrome: from
genetics to pathogenesis". Annual review of genomics and human genetics 2:
271–97.
Piazza, L., Fishman, G., Farer, M., Derlacki, D., & Anderson,
R. (1986). Visual acuity loss in patients with Usher's
syndrome. Archives of Ophthalmology, 104 , 1336–1339.
Plantinga, R., Pennings, R., Huygen, P.,
Sankila, E., Tuppurainen, K., Kleemola, L., et al. (2006). Visual
impairment in Finnish Usher syndrome type III. Acta Ophthalmologica
Scandinavica, 84 (1), 36–41.
Reiners, J; Nagel-Wolfrum, K; Jürgens, K;
Märker, T; Wolfrum, U (2006). "Molecular basis of human Usher syndrome:
deciphering the meshes of the Usher protein network provides insights into the
pathomechanisms of the Usher disease". Experimental eye research 83 (1):
97–119.
Reisser, C., Kimberling, W., & Otterstedde,
C. (2002). Hearing loss in Usher syndrome type II is non progressive. Annals
of Otology, Rhinology and Laryngology,111, 1108–1111.
Roux AF, Faugere V, Le Guedard S, Pallares-Ruiz
N, Vielle A, Chambert S, Marlin S, Hamel C, Gilbert B, Malcolm S, Claustres M
(2006). "Survey of the frequency of USH1 gene mutations in a cohort of
Usher patients shows the importance of cadherin 23 and protocadherin 15 genes
and establishes a detection rate of above 90%". J Med Genet 43 (9):
763–768.
Sadeghi, A., Eriksson, K., Kimberling, W.,
Sjostrom, A., & Moller, C. (2006). Longterm visual prognosis in Usher
syndrome types 1 and 2. Acta Ophthalmologica Scandinavica, 84, 537–544.
Sadeghi, M., Cohn, E., Kelly, W., Kimberling,
W., Tranebjarg, L., & Moller, C. (2004). Audiological findings in
Usher syndrome types IIa and II (non-IIa). International Journal of
Audiology, 43, 136–143.
Sadeghi, M., Cohn, E., Kimberling, W., Tranebjarg, L., & Moller,
C. (2005). Audiological and vestibular features in affected subject with
USH3: A genotype/phenotype correlation. International Journal of
Audiology, 44, 307–316.
Sankila EM, Pakarinen H, Kääriäinen H, Aittomäki
K, Karjalainen S, Sistonen P, de la Chapelle A (1995). "Assignment of
Usher syndrome type III (USH3) gene to chromosome 3q". Hum. Mol. Genetics
4 (1): 93–98.
Schorr, E., Roth, F., & Fox, N.
(2009). Quality of life for children with cochlear implants: Perceived
benefits and problems and the perception of single words and emotional
sounds . Journal of Speech, Language, and Hearing Research,
52, 141–152.
Seeliger, M., Zrenner, E., Apfelstedt-Sylla, E.,
& Jaissle, G. (2001). Identification of Usher syndrome subtypes by ERG
implicit time. Investigative Ophthalmology and Visual Science, 42, 3066–3071.
Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis
RA, et al. (1994). "Clinical diagnosis of the Usher syndromes. Usher
Syndrome Consortium". American Journal of Medical Genetics 50 (1): 32–38.
Steinberg, A., Kaimal, G., Ewing, R., Soslow,
L., Lewis, K., Krantz, I., & Li, Y. (2007). Parental narratives of
genetic testing for hearing loss: Audiologic implications for clinical work
with children and families. American Journal of Audiology, 16, 57–67.
Usher C (1914). "On the inheritance of
Retinitis pigmentosa with notes of cases". Roy. Lond. Ophthalmol. Hosp.
Rep. 19: 130–236.Wagenaar, M., Snik, A., Kimberling, W., & Cremers, C.
(1996). Carriers of Usher syndrome type IB: Is audiometric identification
possible? The American Journal of Otology, 17, 853–858.
Vernon M (1969).
"Usher's syndrome — deafness and progressive blindness. Clinical cases,
prevention, theory and literature survey". Journal of Chronic Diseases 22
(3): 133–151.
von Gräfe A (1858).
"Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der
Netzhaut". Archiv für Ophthalmologie 4: 250–253.
Wallber, J. (1995–2005). Annual reports
of the Eye & Ear Clinic. Rochester, NY: National Technical
Institute for the Deaf at Rochester Institute of Technology.
Williams DS (2007). "Usher syndrome: Animal
models, retinal function of Usher proteins, and prospects for gene
therapy". Vision Research xx (3): xx–xx.doi:10.1016/j.visres.2007.08.015Web
Resources
Boys Town National Research Hospital Usher Syndrome Web page
National Consortium on Deaf-Blindness
National Consortium on Deaf-Blindness, State Deaf-Blind Projects
National Institute on Deafness and Other Communication Disorders
Usher Syndrome Web page
National Institutes of Health Usher Syndrome Web page
National Library of Medicine Genetics Home Reference
Second Sight Medical Products, Inc. retinal
prosthesis—investigational device
Understanding Usher Syndrome: Information for School Counselors [PDF, 2 MB]