Neurinoma dell’acustico
- Categoria: IPOACUSIA NEUROSENSORIALE DELL’ADULTO DI TIPO PROGRESSIVO
- Pubblicato: Martedì, 02 Febbraio 2016 07:37
- Visite: 31072
NEURINOMA ACUSTICO- NEURINOMA DEL NERVO VIII
SCHWANNOMA VESTIBOLARE
NEUROFIBROMATOSI TIPO 2(NF-2)
INDICE
Che cosa è la neurofibromatosi di tipo 2?
Quanto è diffusa la neurofibromatosi di tipo 2? Epidemiologia
Crescita e Cause del Neurinoma
Segni e Sintomi
Quali sono i sintomi di un neuroma acustico?
Perdita Uditiva Acufene Vertigine
Paralisi Facciale
Diagnosi del Neurinoma Acustico :
Audiometria Convenzionale Esami Abr ENG
Immagini Cerebrali
Diagnosi Differenziale
Gestione dei Neuromi Acustici
|
|
|
|
Neurinoma del Nervo Acustico (gonfiore del nervo 8°, appena al di sotto del nervo facciale) |


Punti Principali:
- 1. Il Neuroma del n. acustico rappresenta, una rara causa,di perdita uditiva unilaterale, vertigini, e raramente vi sono altri sintomi cerebrali.
- 2. Le prove migliori per fare diagnosi di neuroma del n. acustico sono gli esami audiometrici e la RM della testa con mezzo di contrasto (gadolinio).
- 3. Circa la metà, di tutti i neuromi acustici , sono trattati con la chirurgia, circa un quarto con radioterapia ( in aumento), ed un quarto controllati nel tempo .
- 4. Non importa quale metodo di trattamento viene utilizzato, la conservazione dell'udito è molto difficile.
CHE COSA È LA NEUROFIBROMATOSI DI TIPO 2?
Ipoacusia genetica associata a disturbi neurologici
Neurofibromatosi tipo 2(NF-2).
- trasmissione autosomica dominante
- elevata penetranza
- neoplasie del sistema nervoso centrale
- neurofibromatosi cutanea
- dicromatismi cutanei
Ci sono due tipi principali di neurofibromatosi:
• Neurofibromatosi di tipo 1 (NF1) è il tipo più comune di neurofibromatosi, che colpisce circa uno su 3.000 persone.
• Neurofibromatosi di tipo 2 (NF2) è meno comune, che colpisce circa uno su 35.000 persone.
|
|
|
|
Neurofibromatosi di tipo 1 (NF1) |
Neurofibromatosi di tipo 2 (NF2) |


Nonostante che condividono lo stesso nome, i due tipi di neurofibromatosi sono distinte condizioni che hanno diverse cause e sintomi.
La Neurofibromatosi tipo 2 (NF-2) è determinata da una mutazione nel gene “merlin”, un gene soppressore tumorale localizzato nella regione 22ql2. Presenta modalità di trasmissione autosomica dominante a penetranza elevata (123). Il 50% dei casi è rappresentato da nuove mutazioni. Questa condizione patologica si ritrova in circa il 5% di soggetti affetti da neurinoma dell’acustico (sordità neurosensoriale retrococleare) Il quadro clinico è rappresentato dalla presenza di tumori del sistema nervoso centrale e periferico (neurinomi, glomi, meningiomi), che a seconda della loro sede danno luogo a sintomi diversi Le prime manifestazioni avvengono in età giovanile, spesso è presente
La Neurofibromatosi di tipo II (o "Sindrome MISME", per "Schwannomi Multipli Ereditati, meningiomi , e Ependimomi ") o Neurofibromatosi centrale, o neurofibromatosi tipo 2 (NF2),è una malattia ereditaria , genetica caratterizzata dalla predisposizione a sviluppare una varietà di tumori del sistema nervoso centrale e periferico. La manifestazione principale della malattia è lo sviluppo di tumori cerebrali non- malignisimmetrici, nella regione del VIII nervo cranico , che è il "nervo cocleo -vestibolare" che trasmette informazioni sensoriali dal orecchio interno al cervello. La maggior parte delle persone con questa condizione possono avere problemi visivi. La NF2 è causata da mutazioni del gene [1] "Merlin" che sembra influenzare la forma e il movimento delle cellule . I principali trattamenti consistono nella rimozione neurochirurgica dei tumori e nel trattamento chirurgico delle lesioni oculari. In contrasto con la neurofibromatosi di tipo 1 (NF1), la NF2 produce una scarsità di manifestazioni cutanee.
Quanto è diffusa la neurofibromatosi di tipo 2? Epidemiologia
La prevalenza alla nascita della Neurofibromatosi tipo ii è stata stimata di 1/40.500- 87.000; circa il 4% dei soggetti con neurinoma dell’VIII nervo cranico presenta una NF’-Z (126, 127, 128). Nel 1920 Feiling e Ward (Evans D.G.R., et AL., 1999) descrissero una famiglia con neurinoma dell’Vili nervo cranico bilaterale ed ipoacusia neurosensoriale, tuttavia casi isolati erano già stati descritti un secolo prima (Feiling A., Ward E., 1920).
L'incidenza stimata di neurofibromatosi tipo 2 (NF2) è di 1 su 37.000 all'anno, con circa la metà degli individui affetti rappresentano i primi casi in famiglia a seguito di nuove mutazioni dominanti.
Sebbene il cambiamento genetico che causa NF2 è presente al momento del concepimento, le manifestazioni cliniche si verificano nel corso di molti anni. L'età tipica di esordio dei sintomi è nella tarda adolescenza a 20 anni, ma la fascia d'età copre l'intero ciclo di vita Goutagny S,.e 2013. Alcune evidenze indicano che l'età di insorgenza dei sintomi clinici è più basso in NF2 maternamente trasmesso. Mentre NF2 è abbastanza variabile in gravità da persona a persona, famiglia studi hanno dimostrato una certa coerenza intrafamilial in età di insorgenza. Mosaicismo somatico per la mutazione NF2 in casi sporadici può anche complicare il quadro clinico, con conseguente sottostima o diagnosi tardiva.
Tuttavia, poiché circa la metà dei casi derivano da nuove mutazioni, la storia familiare è spesso negativa.
Diversamente dal NF1, che spesso è associato con un numero di indizi diagnostici cutanei, l’NF2 è accompagnato da pochi segni esterni. I Sintomi con cui si presenta sono i seguenti:
Nella maggior parte dei casi i sintomi si manifestano nell’adolescenza o attorno ai 20 anni; raramente possono comparire precocemente, durante i primi 10 anni di vita, o tardivamente, dopo i 70 anni. Circa il 70% dci soggetti manifesta un deficit della vista (Vishart J.H.,1822). Dal 20 al 65% dei casi sviluppano pochi piccoli neurofibromi cutanei, soprattutto a livello dello scalpo (Maccollin M., Maumer VF., 1998). Circa il 40% degli affetti presenta poche piccole macchie cutanee color ‘caffelatte” (Kluwe I., Et Al., 2000). La perdita dell'udito, ronzio nelle orecchie, e problemi di equilibrio associata a lesioni del nervo vestibolare Sembra possibile identificare due Forme: il Tipo Wishart con neurinomi dei nervi cocleo-vestibolari e di altri nervi ad insorgenza precoce e decorso rapido (Feiling A., Ward E., 1920), e il Tipo Gardner con neurinomi dei nervi cocleo-vestibolari solamente ad insorgenza tardiva e decorso lento (Kanter W.R., EI AL.,1980; Gardner W.J., Frazier C.H., 1999).
Criteri diagnostici sono considerati: neurinoma dell’Vili nervo cranico bilaterale, un familiare di primo grado affetto da NF-2 e neurinoma dell’VIII nervo cranico monolaterale, oppure nn familiare di primo grado affetto da NF-2 e due delle seguenti forme: glioma, meningioma, neurofibroma, schwannoma, opacità lenticolare subcapsulare posteriore giovanile ( Evans D.G.R., Et AL., 1992) Paralisi dei nervi cranici
I Neuromi acustici, noti anche come neurinomi vestibolari, non sono tumori maligni dell'ottavo nervo cranico . Generalmente nel 98% dei casi prendono origine dalle cellule del rivestimento (cellule di Schwann) del nervo vestibolare inferiore (Komatsuzaki e Tsunoda, 2001; Kras, 2007). Possono anche insorgere nel labirinto (Neff et al, 2003). Insorge, nella maggioranza dei casi,nel nervo vestibolare (superiore o inferiore), in corrispondenza del passaggio tra il rivestimento mielinico centrale e quello periferico, nella così detta ‘transition zone’, dove l’anatomia delle fibre, consistenza della mielina e fine vascolarizzazione intraneurale, sono più fragili. Questa zona di transizione è situata a livello del meato acustico interno. La definizione più corretta sarebbe in effetti schwannoma vestibolare e così è oggi definito nella letteratura internazionale, è chiamato neurinoma dell’acustico per la sintomatologia clinica, che, in una prima fase almeno, è a carico del nervo uditivo (nervo cocleare propriamente detto) per un evidente effetto compressivo e, talora, persino destruente delle fibre uditive da parte delle cellule tumorali. E’ questa sintomatologia che rende diagnosticabile il neurinoma in fasi precoci.
|
Fig.2A-2B Distribuzione dei casi di neurinoma del VIII secondo l’età ed il sesso. A maschi ,B donne (Dutton ed al 1999)
|

Si possono anche produrre nel labirinto (Neff et al, 2003). Essi comprendono circa il 6 per cento di tutti i tumori intracranici, dopo i gliomi e i meningiomi. (Anderson et al, 2000), circa il 30% dei tumori del tronco cerebrale, e circa il 85% dei tumori nella regione ponto-cerebellare - altro 10% sono meningiomi.Il numero 6% probabilmente è troppo alto come i meningiomi e tumori ipofisari sono sottostimati. Solo circa 10 tumori acustici per milione di persone sono ultimamente diagnosticati ogni anno (Evans et al, 2005), corrispondenti a 2000 e 3000 nuovi casi ogni anno negli Stati Uniti. Un altro modo di vedere questo è che una persona media ha un rischio di circa 1/1000 di sviluppare un neuroma acustico nel corso della loro vita (Evans et al, 2005). In Danimarca, l'incidenza annuale è stato stimato a 7,8 pazienti operati / anno (Tos et al, 1992). Come la tecnologia è migliorata, i tumori più piccoli sono stati diagnosticati, con conseguente analoga stima di circa 10 tumori / milioni / anno. Si manifestano prevalentemente nel sesso femminile tra i 35 e 60 anni di età (Fig.2A-2B).
E’ stato descritto per la prima volta da Sandifort E. nel 1772.circa il 30% dei tumori del tronco encefalico, e circa l'85% dei tumori nella regione del ponto-cerebellare - un altro 10% sono rappresentati dai meningiomi. Sono solo circa 10, i nuovi tumori , che vengono diagnosticati ogni anno per milione di persone (Evans et al, 2005), corrispondente negli Stati Uniti a 2000/ 3000 nuovi casi il ogni anno. Un altro modo di considerare questi dati è che una persona normale ha un rischio di circa 1 / 1000 di sviluppare un neuroma acustico nel ciclo della propria vita (Evans et al, 2005).
Nei pazienti con asimmetria uditiva , si ritiene che solo 1 su 1000 abbia una neuroma acustico (fonte: NIH), sebbene alcuni autori riportano una prevalenza più alta 2,5% elevati a (Baker et al.2003). Questa prevalenza più elevata non risulta dalla nostra esperienza clinica. I neuromi acustici si verificano soprattutto negli adulti - sono molto rari nei bambini. Solo in 39 casi di neurinomi nei bambini è stato riportato riportati in letteratura, come ad esempio nel 2001 (Pothula et al, 2001).
Quali geni sono legati alla neurofibromatosi di tipo 2?
Le mutazioni nel gene causa neurofibromatosi di tipo 2 NF2 .Il gene NF2 fornisce istruzioni per fare una proteina chiamata Merlin smeriglio (noto anche come schwannomin). Questa proteina viene prodotta nel sistema nervoso, in particolare nelle cellule di Schwann, che circondano e isolano le cellule nervose (neuroni) nel cervello e nel midollo spinale. Merlin agisce come un soppressore del tumore, il che significa che permette alle cellule di crescere e dividendo troppo rapidamente o in modo incontrollato. Anche se la sua funzione esatta non è nota, questa proteina è probabile anche coinvolto nel controllo dei movimenti delle cellule, forma delle cellule, e la comunicazione tra le cellule. Mutazioni nel gene NF2 piombo per la produzione di una versione non funzionale della proteina esmerejon che non può regolare la crescita e la divisione delle cellule. La ricerca suggerisce che la perdita di merlin permette alle cellule, in particolare le cellule di Schwann, per moltiplicare troppo frequentemente e formare tumori caratteristici della neurofibromatosi di tipo 2.
Per saperne di più sul NF2 gene.
Come fanno i pazienti ad ereditare la neurofibromatosi di tipo 2?
|
|
NF-2 può essere trasmessa come autosomica dominante moda, così come attraverso la mutazione casuale. |

La Neurofibromatosi di tipo 2 è considerato abbia un modello di trasmissione autosomica dominante, anche se la metà degli individui affetti hanno NF2 a seguito di una nuova mutazione del gene.. Le persone con questa condizione sono nati con una copia mutata del gene NF2 in ogni cellula. In circa la metà dei casi, il gene alterato è ereditato da un genitore affetto. I casi rimanenti derivano da nuove mutazioni del gene NF2 e si verificano in persone senza storia di malattia nella loro famiglia. L'incidenza della malattia è di circa 1 a 60.000 Evans DG (2009). Vi è un ampio spettro clinico noto, ma tutti i pazienti controllati sono stati trovati per avere qualche mutazione dello stesso gene sul cromosoma 22 . Attraverso le statistiche, si sospetta che una metà dei casi siano ereditati, e la metà sono il risultato di nuove mutazioni .
A differenza di molte altre condizioni autosomiche dominanti, in cui per causare il disturbo è sufficiente una copia alterata del gene in ogni cellula, due copie del gene NF2 devono essere modificati per innescare la formazione di tumori nella neurofibromatosi di tipo 2 Una mutazione nella seconda copia di il gene NF2 si verifica in cellule di Schwann o altre cellule del sistema nervoso durante la vita di una persona. Quasi tutti coloro che sono nati con una mutazione NF2 acquista una seconda mutazione (noto come una mutazione somatica) in queste cellule e sviluppa i tumori caratteristici della neurofibromatosi di tipo 2.
Patogenesi, Biologia Molecolare e rapporti fisiopatologici
NF 2 è causata da un difetto nel gene che produce normalmente un prodotto chiamato Merlin o Schwannomin, localizzato sul cromosoma 22 banda q11-13.1. Si pensa che Questo peptide abbia una funzione soppressiva sul tumore. In una cellula normale, le concentrazioni di Merlin attivo (defosforilato) siano controllati mediante processi quali l’adesione cellulare (il che indica la necessità di limitare la divisione cellulare). E 'noto che la carenza di Merlin può provocare progressione mediata attraverso il ciclo cellulare a causa della mancanza di soppressione tumorale contatto mediata, sufficiente a causare tumori caratteristici della neurofibromatosi tipo II. Mutazioni di NF II si presume provochino sia un fallimento per la sintesi del Merlin o la produzione di un peptide difettoso che manca il normale effetto soppressivo sul tumore. Il peptide Schwannomin è formato da 595 aminoacidi . Il confronto dello Schwannomin con le altre proteinemostra somiglianze alle proteine che collegano ilcitoscheletro alla membrana cellulare . Mutazioni nel gene-Schwannomin si pensa alterino il movimento e la forma delle cellule colpite con perdita di inibizione da contatto.
Patologia

![]()
Schwannoma del N. Vestibolare

![]()
Meningioma in un paziente con NFII
Il cosiddetto neuroma acustico NF II è in realtà uno Schwannoma del nervo, vestibolare o Schwannoma vestibolare. Il termine improprio di neurinoma del nervo acustico è ancora utilizzato spesso. Gli Schwannomi vestibolari crescono lentamente all'ingresso interna del meato uditivo interno (meato acoustico interno). Esse derivano dalle guaine nervose della parte superiore dei nervi vestibulari nella regione tra la mielina centrale e periferica (Obersteiner-Redlich-Zone) all'interno dell'area del poro acustico, ad 1 cm dal tronco cerebrale.
Correlazione Genotipo-fenotipo
Molti pazienti con NF II sono stati inclusi negli studi che sono stati progettati per confrontare tipo di malattia e la progressione con la determinazione esatta della mutazione associata. Lo scopo di tali confronti di genotipo e fenotipo è determinare se mutazioni specifiche causano rispettive combinazioni di sintomi. Questo sarebbe estremamente utile per la predizione della progressione della malattia e la pianificazione di terapia partendo in giovane età. I risultati di tali studi sono i seguenti:
· Nella maggior parte dei casi peptidi La mutazione nel gene cause NF II abbreviato.
· Non ci sono hot-spot mutazionali.
· I pazienti con mutazione Frameshift - o mutazioni nonsenso soffrono prognosi infausta.
· I pazienti con mutazioni missense hanno una prognosi migliore.
· Nei casi con mutazioni nel splice accettore-regione , non c'è una buona correlazione per determinare.
· Le mutazioni puntiformi possono avere solo effetti minori.
· I casi sono pubblicati [ citazione necessaria ], in cui esattamente la stessa mutazione è associata ad esito chiaramente diverso.
Questi risultati suggeriscono che probabilmente altri fattori (ambientali ed altre mutazioni) determineranno l'esito clinico.
Una mutazione frameshift (chiamato anche un errore di frame o l'orientamento reading frame) è una mutazione genetica causata da indels ( inserzioni o delezioni ) di un numero di nucleotidi in una sequenza di DNA che non è divisibile per tre. A causa della natura tripletta di espressione genica da codoni , l'inserimento o l'eliminazione possono cambiare il reading frame (il raggruppamento dei codoni), risultando in un completamente diversa Traduzione dall'originale.
In genetica , una mutazione non senso è una mutazione puntiforme in una sequenza di DNA che si traduce in un premature codone di stop che provoca il troncamento delle proteine risultanti, o un codone dialogo nel mRNA trascritta e in un tronco incompleto e di solito prodotta proteina non funzionale
In genetica , una mutazione missenso (un tipo di sostituzione nonsynonymous ) è una mutazione puntiforme in cui un singolo nucleotide risulti cambiamento in un codone che codifica per un diverso amminoacido . [1]
CRESCITA E CAUSE DEL NEURINOMA
Le cause che determinano la crescita di un neurinoma non sono attualmente note . Circa 7 su 100 neuromi acustici sono causati da neurofibromatosi tipo 2 (NF2). NF2 è una malattia genetica molto rara che provoca tumori benigni del sistema nervoso. Essa colpisce circa 1 su 350.000 persone. Quasi tutti con NF2 sviluppa un neuroma acustico su entrambi i nervi acustici (cioè, tumori bilaterali). Il neurinoma del nervo acustico si presenta in due forme: una forma sporadica ed una forma associata con una sindrome ereditaria chiamata neurofibromatosi di tipo II (NF2). Circa il 93 per cento di tutti i casi sono sporadici.
Maggiori informazioni su NF2 si possono trovare alla http://ghr.nlm.nih.gov/gene=nf2
Attualmente non vi è evidenza che le radiazioni emesse dai telefoni cellulari possono causare neuroma acustico (Muscat et al, 2002). Solo nei casi di Neurofibromatosi tipo II si può attribuire una causa genetica. I tumori del nervo acustico generalmente hanno una crescita lenta (circa 2 mm). Se il neurinoma, come avviene nella maggior parte dei casi, si sviluppa nel segmento del nervo in prossimità delineato acustico interno, e pertanto può espandersi (la sua crescita è di ca. 0,5 cm. all’anno) verso il meato acustico interno e attraverso questo, diffondersi nella cavità endocranica .A tal punto il quadro clinico si modifica. Il deficit uditivo peggiora. si aggravano la vertigine, i disturbi dell’equilibrio e la sensazione di incertezza della marcia. Compare la cefalea che e prevalentemente localizzata in sede occipitale o frontale Si rendono manifesti i segni di sofferenza del n. faciale a causa della comparsa dell’asimmetria della rima palpebrale e di quella buccale. Essi rimangono avvolti da una capsula e tendono a erodere l'osso e a spostare i tessuti neurovascolari normali. Per la crescita lenta esiste un adattamento graduale dell'organismo alternato a periodi di sintomi più o meno evidenti dovuti alla compressione del nervo uditivo e/o del nervo facciale, quindi il tumore esce dal canale osseo ed invade lo spazio dell'angolo ponto-cerebellare. A questo stadio, alla risonanza magnetica cerebrale, il tumore assume l'aspetto di una massa globosa peduncolata sul C.U.I.. Nella sua ulteriore crescita, il tumore comprime il nervo trigemino ed il tronco cerebrale rendendo il trattamento chirurgico non dilazionabile.
|
|
|
Figura 2: Neurinoma dell'acustico sinistro di circa 1,5 cm nell'angolo ponto-cerebellare. Non compressioni sul tronco cerebrale. Il CUI non appare deformato ed è appena lievemente allargato. Figura 3: Neurinoma dell'acustico destro di dimensione superiore a 4 cm. Importante deformazione del tronco cerebrale e del cervelletto. Notevole allargamento del CUI
|

|
|
|
Figura 3: Neurinoma dell'acustico destro di dimensione superiore a 4 cm. Importante deformazione del tronco cerebrale e del cervelletto. Notevole allargamento del CUI |

Compare la cefalea che e prevalentemente localizzata in sede occipitale o frontale Si rendono manifesti i segni di sofferenza del n. faciale a causa della comparsa dell’asimmetria della rima palpebrale e di quella buccale.
Si osserva una riduzione del riflesso corneale e un’alterazione della lacrimazione nell’occhio omolaterale alla sede della lesione. La presenza, nel 25-30% dei casi, di una ipoestesia all’emifaccia indica una sofferenza del V. La comparsa di un deficit a carico di altri nervi cranici (VI, IX, X, XII) dimostra che la neoplasia si è ulteriormente diffusa. Si realizza in tal modo, un quadro clinico neurologico con un corredo sintomatologico multiforme a causa non solo della paralisi dei nn. cranici e dell’interessamento dei distretti bulbare, cerebellare e pontino ma anche della comparsa di un’ipertensione endocranica per l’ostacolo al deflusso del liquor.
SEGNI E SINTOMI:
La diagnosi di una richiede di solito un medico con esperienza otologica che possono integrare insieme l'intera immagine, o una risonanza magnetica con gadolinio(Fig. 8d ). Perché neuromi acustici sono molto rari, e la RM sono molto costose, a nostro parere - tutti i pazienti con un rischio sostanziale di avere una chitarra acustica devono essere valutati da un medico con esperienza otologica. In altre parole , tutti i pazienti , con una inspiegabile stabile perdita uditiva, asimmetrica , in generale dovrebbero essere valutati da un medico con esperienza otologica. Il testo seguente descrive come questo processo di integrazione possa essere effettuato .
Quali sono i sintomi di un neuroma acustico?
I sintomi iniziali sono costituiti da ipoacusia, che si verifica in 9 su 10 pazienti, cioè diminuzione progressiva dell’udito dal lato della neoformazione che spesso inizia con un’alterata comprensione del significato di parole e frasi nella conversazione al telefono, da acufeni (ronzii e fischi all’orecchio). Gli acufeni, che si presenta in 7 su 10 persone, rappresentano il motivo più frequente di visita medica in questi pazienti. A questi due sintomi spesso si associa un lieve disturbo dell’equilibrio, quasi la metà delle persone con un neuroma acustico hanno questo sintomo, ma meno di 1 su 10 lo hanno come primo sintomo. Intorpidimento del viso, formicolio o dolore . Questi sintomi sono dovuti alla pressione del neuroma acustico su altri nervi. Il nervo comunemente colpite è chiamato il nervo trigemino che controlla sensazione in faccia. Circa 1 persona su 4 con neurinoma acustico ha qualche intorpidimento del viso - questo è un sintomo più comune di quanto la debolezza dei muscoli facciali. Tuttavia, è spesso un sintomo inosservato ; mentre la perdita dell'udito è comune nel neuroma acustico (vale a dire che è sensibile), ci sono miriadi di altre cause di perdita dell'udito (l’ipoacusia cioè è molto aspecifica). A causa della elevata sensibilità, ma bassa specificità, l'uso di routine di un test diagnostico molto costoso come un gad-RM(risonanza magnetico con gadolinio) della IAC(condotto uditivo interno) in tutte le persone con una perdita uditiva asimmetrica non è sempre giustificata. In altre parole, gli errori sono possibili sulla base del rapporto costi / benefici sociali. Mentre alcuni clinici effettuano una RM su tutti i pazienti con udito asimmetrico, questo è un modo molto costoso per "trovare" una neurinoma acustico.
La perdita dell'udito è il sintomo più frequente del neuroma acustico, che si verificano in più del 95% dei pazienti. L'ipoacusia è generalmente associata ad un ronzio o rumore monolaterale nonchè a disturbi dell'equilibrio con instabilità o, raramente, vere crisi vertiginose. Non è raro che un neurinoma dell'acustico possa presentarsi con i sintomi di una Malattia di Ménière (vertigini violente accompagnate da perdita di udito e acufeni).. Circa il 90 per cento si presentano con una , perdita uditiva lentamente progressiva monolaterale. Un esempio è riportato qui di seguito.
I medici spesso cercano di stimare il rischio di un neuroma acustico guardando il modello di perdita dell'udito (vedi sotto per audiogrammi più frequenti). Il tipo più comune è un pattern neurosensoriale ad alta frequenza, si verifica in circa in due terzi dei pazienti. Nel terzo rimanente l'osservazione più comune è una perdita dell'udito a bassa frequenza (che sarebbe più tipico della Malattia di Ménière). Ancora meno frequentemente, alcuni hanno il morso "cookie pattern " (indicativi di perdita dell'udito congenita o di una incisura da rumore noise notch).
Alcune volte il sintomo rilevatore di un neurinoma dell'acustico può essere una sordità improvvisa, talvolta con recupero uditivo dopo trattamento medico, oppure il paziente può sperimentare una ipoacusia fluttuante o senso di orecchio pieno. Una perdita uditiva improvvisa si verifica in circa il 25 per cento dei pazienti con neuroma acustico. Tuttavia, poiché il neuroma acustico è una condizione rara, la perdita uditiva improvvisa dovuta a un tumore acustico si verifica solo nel 1-5 per cento dei pazienti con perdita uditiva improvvisa in quanto ci sono molte cause più comuni (Daniels et al, 2000). Anche una perdita improvvisa dell'udito, con recupero completo può essere causato da una chitarra acustica(Nageris e Popovtzer, 2003).
Una ipoacusia asimmetrica è sensibile ma non specifica di neuroma acustico. La mancanza di specificità e rarità di neuromi acustici, rispetto alla miriade di altre cause di audizione asimmetrica rende il "costo" della scansione di ogni persona con un udito asimmetrico, per trovare una chitarra acustica in 1 / 1000 estremamente elevato. La mancanza di specificità è stato commentato da Margolis e Saly (2008). Questa conclusione deve essere temperata da altre informazioni cliniche ,qualcuno con una progressiva riduzione asimmetrica dell'udito neurosensoriale avrà (a nostro parere) una probabilità di gran lunga maggiore, di avere una chitarra acustica rispetto a qualcuno con una asimmetria statico o che migliora nel tempo .
La specificità è un'altra considerazione è. A questo proposito, alcuni riscontrano una funzionalità uditiva del tutto normale in circa l'11% dei pazienti (Morrison e Sterkers, 1996). A parere, di Heine tale percentuale è alta, ma certamente il neurinoma dell’acustico si può trovare in persone con udito simmetrico.
Acufene
L’Acufene è molto comune nel neuroma dell’acustico, è solitamente unilaterale e limitato nell'orecchio colpito, se avete un acufene, è molto molto improbabile che si abbia un neuroma acustico, perché questi tumori sono molto più rari rispetto ad altri meccanismi di danno uditivo.
Vertigine
Nonostante l'origine abituale del neurinoma acustico dal nervo vestibolare inferiore (Komatsuzaki e Tsunoda, 2001; Kraj et al, 2007), le vertigini (spinning) prima di un intervento chirurgico non sono comuni, si verificano solo in circa nel 20 per cento delle persone con neuroma acustico. Quando la neoplasia si espande nell’angolo ponto-cerebellare a ridosso delle strutture nervose centrali, compare un nistagmo nello sguardo diretto verso il lato della lesione (nistagmo di Bruns) E di tipo orizzontale, aritmico. con scosse di piccola ampiezza. In questo stadio del neurinoma, si osservano quindi due tipi di nistagmo di opposta direzione l’uno spontaneo di tipo “deficitario”, presente fin dall’inizio della malattia dovuto all’asimmetria vestibolare periferica, l’altro “da direzione dello sguardo”, di tipo “irritativo”, omolaterale alla sede della lesione d’origine centrale, Questo nistagmo indica che la neoplasia. dopo essersi estesa all’angolo ponto-cerebellare, comprime parzialmente le aree nucleari vestibolari del bulbo e contemporaneamente le vie di connessione vestibolo-cerebellari omolaterali alla sede della neoplasia (Fig. 4A).
Anche la motilità oculare riflessa risulta compromessa. Si osserva, infatti, una anomalia dei movimenti lenti di inseguimento (pursuit) diretti verso il lato del neurinoma. Il NOC risulta facilitato, per l’interferenza del nistagmo spontaneo quand’è diretto verso il lato opposto alla sede del tumore, mentre appare parzialmente inibito e aritmico quand’ è diretto omolateralmente
A causa dell’ulteriore estensione del tumore, l’interessamento delle strutture nervose del tronco encefalico si fa più evidente. Il nistagmo da “direzione dello sguardo” omolaterale alla sede del tumore subisce delle profonde modificazioni del ritmo e della forma delle scosse. Se si invita il paziente a volgere lo sguardo verso il lato malato. si nota infatti che le scosse nistagmiche appaiono, all’inizio, assai frequenti ma. dopo qualche secondo, esse si riducono progressivamente di ampiezza e di numero man mano che i bulbi oculari ritornano lentamente e in modo del tutto involontario nella posizione primaria di sguardo.
|
|
Fig. 4A - Z.S., a. 52 - Neurinoma dell’VIII° di destra. Ipoacusia neurosensoriale destra, nistagmo spont. “deficitario” diretto verso sinistra; nistagmo di Bruns diretto verso destra; iporeflettività vest. destra allo stim. cal. (20°) del lab.; RMN Con mdc evidenzia una formazione ovalare a Contorni netti, ancora parzialmente insinuata nel meato ac. int. Che determina modesta impronta sul ponte e sull’emisfero cerebellare. BABIGHIAN-OTONEURGIA-PICCIN-2008-
|

Si tratta del nistagmo “dello sguardo paretico” che indica una soffe- renza più profonda delle connessioni cerebellopontine e dei centri nervosi del tronco encefalico che regolano l’attività oculomotoria, Una volta che i bulbi oculari hanno raggiunto la posizione primaria dello sguardo. il nistagmo dello “sguardo paretico” cessa totalmente e sul grafico ricompare il nistagmo “deficitario” diretto verso il lato opposto alla sede della lesione (Fig. 4C-D). Il NOC evocato ipsilateralmente alla sede della neoplasia appare, in questo caso, maggiormente alterato e, a volte,può anche mancare. È lo stadio della malattia in cui, oltre alla cefalea occipitale e frontale e agli altri sintomi neurologici (paralisi di nervi cranici, atassia, disdiadococinesi), si osservano i primi segni dell’ipertensione endocranica (papilla da stasi, vomito mattutino a stomaco vuoto, ecc.) mentre i caratteri istochimici del liquor risultano normali. In qualche caso. si può anche notare un nistagmo “alternante” (Fig. 4B) di tipo orizzontale o diagonale che, come si è già detto, è caratterizzato da gruppi di scosse di opposta direzione che si alternano tra loro separati da brevi periodi di pausa Secondo la maggior parte degli Autori, tale nistagmo dipenderebbe dalla mancanza dell’azione inibitrice che le aree cerebellari svolgono sui neuroni dei nuclei vestibolari mentre per altri il nistagmo “alternante” conseguirebbe all’interessamento di alcune strutture nervose del tronco encefalico in particolare della reticolare paramediana del tetto del ponte e anche dei mesencefalo.

Fig. 4B nistagmo “alternante”
La diagnosi differenziale andrà posta con alcun neoplasie della fossa cranica posteriore: meningioma, lipoma. angioma, cisti aracnoidea, tumore del giorno giugulare, neurinoma di altri nervi cranici, medulloblastoma o con altre lesioni quali l’aracnite della base. anomalie vasali, etc. Anche le metastasi di neoplasie del polmone, del rene, della prostata e della mammella che realizzano la cosiddetta “sindrome maligna dell’angolo ponto-cerebellare” dovranno venir considerate per la diagnosi differenziale.
|
|
Fig. 4C- F.S, a. 22. Neurinoma dell’VIII° in sede di a. ponto-cerebellare destro con paresi del VII. Grave deficit uditivo all’or, destro, nist. spont. “deficitario” diretto verso sinistra; nistagmo dello “sguardo paretico” verso destra (col ritorno involontario degli occhi nella posizione primaria compare il nistagmo spontaneo), areflessia vest. destra. allo stim. cal. (20°) del lab. (A); NOC oriz. alterato; RMN eseguita con mdc rivela una masserella rotondeggiante a livello del meato. ac. mt. di destra che sporge nell’a. pontocerebellare (B). BABIGHIAN-OTONEURGIA-PICCIN-2008-
|

Siccome il nervo vestibolare inferiore innerva il canale semicircolare posteriore ed il sacculo, ci si potrebbe aspettare che i VEMP’s , che sono un esame funzionale del sacculo , siano uniformemente anormali nel neurinoma dell’acustico ed in effetti questi esami sono abbastanza sensibili. Allo stesso modo, ci si potrebbe aspettare che una VPPB ipsilaterale sia rara. Tale questione non è stata affrontata. Si può anche prevedere anomalie nelle OAE in quanto lei vie uditive efferenti entrano nell'area cocleare tramite la divisione inferiore del nervo. Ancora una volta, la questione non è stata affrontata.
|
|
Fig. 4D Neurinoma dell’VIII° in sede di ang. ponto-cerebellare di sinistra. Anacusia sin.; nist. “deficitario” verso destra, nist. dello ‘sguardo paretico” verso sinistra; areflessia vest, sinistra alla stim. cal. (200) del lab; alterazioni del NOC orizzontale in particolare di quello verso sinistra; RMN con mdc: grossa formazione a livello dell’ang. ponto-cerebellare sinistro. ABR tracciato destrutturato a sinistra, non deficit di altri nervi cranici; non segni di stasi al fundus. BABIGHIAN-OTONEURGIA-PICCIN-2008-
|

La vertigine è più comune nei piccoli tumori .L’instabilità è molto più diffusa rispetto alle vertigini alle vertigini e circa il 70 per cento dei pazienti con tumori di grandi dimensioni hanno questo sintomo. I sintomi CerebellarI (cioè scarso coordinamento delle armi) sono inusuali.
L’iperventilazione che produce nistagmo è un segno fisico poco conosciuto che può essere molto più specifici per il neuroma acustico. Valutazione delle HVIN richiede delle attrezzature più sofisticate di quanto non sia disponibile nella maggior parte degli uffici. Richiede anche che l'esaminatore abbia ad avere familiarità con questo segno -.
Paralisi facciale
I Disturbi Sensoriali Facciali si verificano solo nei tumori di grandi dimensioni (circa nel 50 per cento di quelli con dimensioni superiori a 2 centimetri di). Il disturbo sensoriale del viso può rispondere alla carbamazepina o oxcarbamazine farmaco utilizzato per le nevralgie. Una debolezza del facciale è rara. Contrazioni del viso, note anche come synkinesis facciale o spasmo facciale, si verifica in circa il 10 per cento dei pazienti. Mal di testa prima di un intervento chirurgico si verifica in circa il 40 per cento dei pazienti i con un tumore di grandi dimensioni. . La presenza, nel 25-30% dei casi, di una ipoestesia all’emifaccia indica una sofferenza del V. La comparsa di un deficit a carico di altri nervi cranici (VI, IX, X, XII) dimostra che la neoplasia si è ulteriormente diffusa.
DIAGNOSI DEL NEURINOMA ACUSTICO :
Diagnosi
Prenatale
Neuromi acustici bilaterali sono diagnostici di NF2. [2]
Postnatale
Ferner et al, 2011. danno tre serie di criteri diagnostici per NF2:
1. Schwannoma vestibolare bilaterale (VS) o storia familiare di NF2 più VS unilaterale o due di: meningioma, glioma, schwannoma, neurofibroma, posteriore subcapsulare opacità del cristallino
2. Schwannoma vestibolare unilaterale VS più eventuali due di meningioma, glioma, schwannoma, neurofibroma, posteriore subcapsulare opacità del cristallino
3. Due o più meningioma più VS unilaterale o due di glioma, schwannoma e cataratta.
NF II può essere diagnosticata con il 65% di precisione prenatale con villocentesi o amniocentesi . [4]
Un'altra serie di criteri diagnostici è il seguente:
· Rilevamento di neuroma acustico bilaterale da parte di procedure di imaging
· Parente di primo grado con NF 2 ea comparsa di :
· Neurofibroma , meningiomi , glioma o Schwannoma
· Parente di primo grado con NF 2 e l'insorgenza di cataratta sottocapsulare posteriore giovanile.
La diagnosi di NF2 comporta quanto segue:
I test genetici
Studi di imaging
Esami uditivi, oftalmici, e istologiche
I test genetici
Una volta che la diagnosi clinica è stata stabilita inequivocabilmente in un dato individuo, analisi molecolare diretta può essere offerto
Tassi di rilevamento per i test molecolari basati su approcci 65%
Se viene trovata una mutazione, altri familiari asintomatici possono beneficiare di test presintomatico
Test molecolare del tessuto tumorale può aumentare studi molecolari tradizionali quando l'analisi del DNA ottenuto da linfociti di sangue è non diagnostico
Per le famiglie in cui nessun mutazione può essere identificato in una persona colpita nota, analisi di linkage o metodi di test genetici indiretti possono essere utilizzati
Test presintomatico dei membri a rischio della famiglia richiede un vigoroso processo di consenso informato e potrebbe essere meglio fatto durante una sessione di consulenza genetica in un cancro, centro genetico, o neurofibromatosi specializzata in tali questioni
Test prenatale per NF2 è la seguente:
Quando un genitore ha NF2, test prenatale può essere fatto su amniociti o villi coriali, sia attraverso l'analisi diretta mutazione del gene quando è stata identificata la modifica o mediante analisi di linkage
La diagnosi prenatale può non essere possibile se il genitore affetto è la prima persona colpita in famiglia e una mutazione non può essere trovato
Se un genitore prospettico ha una nota NF2 mutazione, la diagnosi genetica preimpianto può essere possibile se la coppia è disposto a sottoporsi a fecondazione in vitro con trasferimento di embrioni non affetti
La risonanza magnetica
MRI rimane il fondamento per la diagnosi e lo screening del sistema nervoso centrale, nervi cranici e tumori del midollo spinale
Soggetti a rischio possono essere monitorati per i tumori del sistema nervoso centrale che iniziano durante l'adolescenza, con scansioni MRI annuali della testa eseguita attraverso i loro fine degli anni '50
MRI con 3 dimensioni (3D) volumetria è ora il metodo preferito per seguire la crescita schwannoma vestibolare nel tempo Harris GJ, e 2008.
Routine MRI del midollo spinale probabilmente non è indicato nei soggetti asintomatici affetti oa rischio
Risonanza magnetica della colonna vertebrale è indicata diagnostico quando un individuo si presenta con motori o sensoriali modifiche suggestivi di una lesione del midollo spinale o lesioni
Come accennato in precedenza - il neurinoma acustico può essere diagnosticata o da un medico con esperienza otologica, che possa integrare insieme l'intera immagine, o con una risonanza magnetica con gadolinio del cervello. Siccome le MRI, sono molto costose , il metodo più efficiente per contenere i costi della diagnosi (già scritto nella 2008), è che , tutti i pazienti con una inspiegabile perdita dell'udito asimmetrica, siano valutati da un medico con esperienza otologica. Qualcosa che finora è stato trascurato nella valutazione preoperatoria dei pazienti con neurinoma dell’acustico è stato di stabilire preoperatoriamente, in tutti i casi, i parametri della funzione facciale ricavati dalla elettroneuronografia e dal blink test e i dati dell’udito preoperatorio da accertamenti più approfonditi che comprendano:
• es audiometrico tonale e vocale
• test sopraliminari
• audiometria automatica
• potenziali evocati uditivi
• test audiometrici vocali competitivi
• valutazione protesica sia con protesi acustiche tradizionali che con protesi ad induzione magnetica
solo la combinazione di tutte queste informazioni ottenute bilateralmente potrebbe darci la possibilità di offrire informazioni più precise ai nostri pazienti cui va peraltro spiegato che ogni intervento chirurgico è una realtà a sé e che le possibilità offerte sono solo quelle su base statistica (se siamo in due e io mangio una mela per la statistica ne abbiamo mangiato ciascuno il 50%!!!) è inoltre da considerare tra le altre cose anche l’applicazione di un impianto BAHA nei casi di perdita totale dell’udito e, in un futuro si spera non lontano, un impianto cocleare più sofisticato di quelli attualmente disponibili per offrire la possibilità di un ripristino seppur parziale di una funzione uditiva nel lato
|
Fig.5 Audiogramma tipico di paziente con un neuroma acustico. |

Audiometria Convenzionale è il test più utile per diagnosticare un neurinoma acustico. L'anomalia più comune è una perdita neurosensoriale asimmetrica alle alte frequenze (vedi figura 5 sopra). Non più di 1 su 20 pazienti , con tumori di grandi dimensioni , hanno una asimmetria entro 15 dB a 4000 Hz. Tuttavia, bisogna ricordarsi che solo 1 su 1000 pazienti con asimmetria uditiva può avere neuroma del nervo acustico . È stato stimato che il 5 per cento delle persone con ipoacusia neurosensoriale hanno un neurinoma acustico (Daniels et al, 2000), ma questa stima è sospetta in quanto comporterebbe una prevalenza molto più elevato di neuromi acustici di quanto siano comunemente accertati. L'audiometria vocale (SRT) è normale in molti pazienti con tumori di piccole dimensioni. La discriminazione vocale è eccellente in circa il 50% dei pazienti con piccoli tumori, e un terzo dei pazienti con tumori di grandi dimensioni hanno una discriminazione vocale ancora quasi normale (> 80%), la discriminazione discorso. Perché l’asimmetria uditiva è dovuta dovuto principalmente a condizioni diverse da neuromi acustici, altri elementi di informazione devono essere integrati per fare la diagnosi clinica di neuroma acustico. Di solito questo processo di integrazione è fatto da un medico con esperienza otologica, e non dagli audiometristi .
|
|
|
Fig. 6 Audiogramma del paziente con un grande neuroma acustico sul lato sinistro, ma (quasi) la funzionalità uditiva è quasi simmetrica . Questo esempio dimostra che i test con simmetria uditiva, non sempre escludono la diagnosi di un neuroma acustico. Si veda il commento in testo. |

Ipoacusie simmetrica o anche normale funzionalità uditiva non escludere una chitarra acustica, ma è molto raro .si sono trovati diversi pazienti con una funzionalità uditiva simmetrica , ma con un grande neurinoma acustico da un lato sopra è riportato un esempio di un uomo che aveva una grande neurinoma acustico sul lato sinistro - per essere sicuri che si deve fare una risonanza magnetica, è di essere il più sicuri possibile, si deve fare una risonanza magnetica ad alta risoluzione del IAC (condotto uditivo interno )con gadolinio. Perché quest’esame è terribilmente costoso (lo screening di tutti i pazienti con problemi d'udito di qualsiasi tipo), tuttavia a volte sono stati fatti degli errori i. Mentre questa è davvero una decisione che per gli economisti sanitari, ci sembra che complessivamente sono ammissibili degli errori occasionali è ammissibile, se uno considera ciò che è meglio per una grande popolazione .
Esami ABR Fig. 6b
Quando l’esame audiometrico è anormale con un peggioramento uditivo progressivo, audiometria in genere vengono effettuati ulteriori test, come l’ABR (Auditory Brainstem Response ) o risposte uditive del tronco cerebrale, e RM (risonanza magnetica), con determina la diagnosi. L’esame ABR è meno sensibile di quello RM (il tasso di falsi negativi è di circa il 33%), ma è molto meno costosa. Una nuova tecnica chiamata "summated ABR", in sostanza, ABR diversi registrati in tempi diversi , possono dare una migliore sensibilità. Nel nostro contesto clinico, questa tecnica è non è poco indicata e comunemente il test più sensibili per l'acustica (MRI) è così. Tuttavia, questo metodo potrebbe avere utilità in contesti in cui le RM sono di difficile accesso.
Una configurazione caratteristica ABR in una persona con un neuroma acustico è un'onda i e dopo nulla assenza delle onde 3° o 5° (10-20% dei casi). Un ritardo dell’intervallo onda I- III è comune, un ritardo dell’onda V si verifica nel 40-60% dei casi. Con l’ ABR si hanno un elevato numero di falsi positivi e falsi negativi. Fino a 1 / 3 dei pazienti con tumori di piccole dimensioni (con MRI) hanno un ABR normale.
|
|
Fig 6b. ABRs registrato da pazienti con tumori acustici chirurgicamente confermati. La forma d'onda superiore in ogni pannello è da un orecchio normale. La forma d'onda inferiore a (a) mostra l'intervallo I-V prolungato da un paziente con un neuroma acustico. La forma d'onda inferiore a (b) mostra l'assenza dell’ onda V da un paziente con un meningioma dell'angolo ponto-cerebellare. |

Esami Videonistagmografici( ENG)
La Videonistagmografia, (test ENG) è spesso alterata e circa il 60 per cento di tutti i tumori , sono associati ad una iporeflettività unilaterale alle prove caloriche (Hulshof et al, 1989). Tuttavia, l’ ENG non è un test diagnostico specifico. Il Test con la sedia rotatoria è meno sensibile del test calorico. La posturografia è insensibile al neuroma acustico. Il decay test,o test di Anderson (decadimento del riflesso acustico) è anche poco sensibile (circa il 36%). Anche le Otoemissioni acustiche sono considerate un test poco sensibile per il neurinoma acustico. Come accennato in precedenza, ci si aspetterebbe che i VEMP’s siano sensibili al neuroma acustico.
|
|
|
|
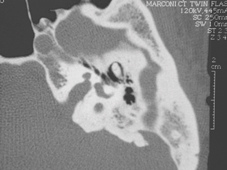
Fig.-7a Risonanza magnetica del cervello (coronale) che mostra un neuroma acustico (la macchia bianca sul lato sinistro della foto). |
Fig.-7b Risonanza magnetica del cervello assiale (con contrasto) che mostra un gran neuroma acustico intracanalicolare, sul lato destro del cervello (parte sinistra della scansione). |
|
|
|
|
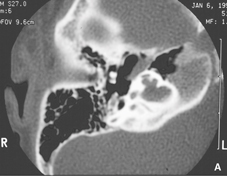
Fig.-7c Un altro neurinoma acustico intracanaliculare sul lato destro. |
Fig.-7d Grande neurinoma acustico di tre centimetri con interessamento del tronco cerebrale sul lato L. |




Esame oftalmico
I controlli annuali sono raccomandate per i bambini e gli adulti con NF2
Visite oculistiche dilatati per opacità del cristallino, amartomi retinici, o membrane epiretiniche può essere molto utile anche in un bambino a rischio di NF2
Cataratta giovanile possono essere visti molto prima che un bambino mostra alcun segno di schwannoma vestibolare
IMMAGINI CEREBRALI
Anche se è relativamente costosa rispetto all’ audiometria o ABR, la prova ottimale per escludere il neuroma acustico è un RMI T1 con gadolinio ( vedi foto sopra). Con la RM, i neuromi acustici sono spesso ingrossati n modo uniforme , densi, e si espandono nel condotto uditivo interno. A Una variante della RM(fast spin-echo T2 )è molto sensibile al neuroma del nervo acustico, e in alcune situazioni cliniche, può essere effettuato a buon mercato. Se una risonanza magnetica non può essere fatta, come ad esempio nei pazienti con un pacemaker o di clips metalliche, - dovrebbero essere effettuata nei soggetti ad alto rischio un CT aperta, in particolare se l'ABR è indicativo di un neuroma acustico. La TAC con contrasto non è un buon esame per la diagnosi di neuromi acustici, in quanto ha un alto tasso di falsi negativi (circa il 37%).
Mentre la RM è la prova più sensibile per il neuroma acustico, anche se si possono commettere errori. Falsi negativi (errori )si verificano soprattutto nelle persone con tumori molto piccoli, o nelle scansioni eseguite male (come ad esempio in una scansione fatto con una bassa-unità di campo e senza contrasto). Errori di tipo falsi positivi sono molto rari, ma anche possibili (House et al, 2008).
Un Neuroma acustico può raggiungere dimensioni anche di a 4 cm. Il più piccolo, neuroma acustico intracanalicolare (vedi sopra a destra), è misurata in millimetri. Un "piccolo" neuroma acustico è inferiore a 1,5 cm (sopra a sinistra). Un neuroma acustico "moderato" acustica è 1,5-3 cm, e un "grande" un neuroma acustico è di 3 cm o superiore.
I tumori sono stadiati in base alla combinazione della loro localizzazione ed alle dimensioni. Un tumore "intracanalicolare" è piccolo ed è collocato nel IAC (condotto uditivo interno). A tumore "cisternale" si estende al di fuori della IAC. Un tumore "compressivo" l è quello a contatto con il cervelletto ed il tronco encefalico, e un tumore "idrocefalico" è quello che sta ostruendo le vie di drenaggio del CSF(liquido-cefalo-rachidiano) nel IV° ventricolo .Il Neuroma acustico si può estendere dal nervo all'orecchio interno,il che può rendere più difficile la loro rimozione (Falcioni et al, 2003). Gli schwannomi Intralabirintici nonché gli schwannoma intracocleari esistono (Kennedy et al, 2004). Il neurinoma acustico riportato al di sopra ,nella figura a destra ha una piccola estensione intracocleare.
|
|
|
Fig.8a Immagine Assiale di un paziente con NF2 mostra un neuroma acustico su entrambi i lati (Image cortesemente fornita dal Dr. Richard Wiet ). Fig.8b Immagine a più elevata definizione che mostra meningiomi multipli in uno stesso paziente con NF2. |

Raramente, i neuromi acustici sono di origine ereditaria. Neuroma acustico causato da una neurofibromatosi di II tipo ,dovrebbe essere sospettata nei giovani pazienti e in quelli con una storia familiare di tumori neurali. La figura sopra mostra un esempio di tale paziente. E 'comune in questa malattia diventare sordi a causa di neuromi acustici bilaterali. Il test genetico per la NF1 e NF2 è disponibile in commercio, per esempio, dalla diagnostica Athena.
|
|
Fig.8c Caratteristiche radiologiche di neuroma acustico: 1. Ampliamento del meato acustico interno "segno di Trumpet" 2. Estensione extracanalicolare ponto-cerebellare (percorso di minor resistenza) può portare a comparsa del "cono gelato” |

|
|
|
Fig.8d MRI petrous bone with gadolinium is the most diagnostic one. |

Diagnosi Differenziale
Ci sono diversi altri tumori che possono verificarsi nella stessa regione del cervello, l'angolo cerebello-pontine o CPA, come i neuromi acustici. Di tutte le lesioni del CPA, l'angolo ponto cerebellare i neuromi acustici rappresentano rappresentano all’incirca il 70-90 per cento. I meningiomi sono secondi per frequenza (10 per cento), seguita dai tumori epidermoidi, e successivamente dai lipomi. Di tanto in tanto i tumori collocati in altri organi, come il polmone, possono metastatizzare al CPA. I tumori metastatici di solito crescono rapidamente – La funzionalità uditiva peggiora velocemente, e spesso il nervo facciale è coinvolto con una paralisi di Bell (Bells palsy), , nel corso di poche settimane.
Neuromi Acustici Scoperto per Caso Accidentalmente
Siccome la TC e la risonanza magnetica sono più comunemente utilizzati, sempre più Neuromi Acustici, sono scoperti casualmente . Per esempio, una persona che ha un mal di testa o emicrania, può richiedere una risonanza magnetica, che rivela un neuroma acustico. O qualcuno che ha avuto un incidente automobilistico, potrebbe richiedere una risonanza magnetica, che rivela un neuroma acustico.
Questa situazione ha un considerevole potenziale di guai. Non è molto probabile che un neuroma acustico in precedenza in silenzio, all'improvviso si manifesta al tempo stesso come un altro problema -, come un lieve infortunio testa o emicrania. Piuttosto, è comunemente il caso che la persona che avrà una delle cause più comuni di vertigine, e appena capita di avere una chitarra acustica in silenzio.
Se qualcuno in una simile situazione procede con la chirurgia o radioterapia, n è certo che l'intervento chirurgico o radiazioni creeranno uno squilibrio vestibolare e sordità. Questo alla fine si verificano in ogni caso, ma il trattamento di un tumore che non è in crescita accelererà il processo. Per questo motivo, estrema cautela è suggerita - a nostro parere, fatta eccezione per i tumori di grandi dimensioni, è meglio avere una prova oggettiva - ossia la progressione della perdita di udito o di un allargamento su MRI - che il tumore è in crescita prima di imbarcarsi su un intervento chirurgico o radioterapia. Vale la pena di chiedere un secondo parere .
I rischi e le complicanze della chirurgia di un tumore del nervo acustico, variano a seconda delle dimensioni.
Più grande è il tumore, più seria e maggiore è lo probabilità di avere delle complicanze. La rimozione di un tumore del nervo acustico, sia grande che piccolo, è un procedimento chirurgico importante , con la possibilità di gravi complicazioni, inclusa la morte.
Il rischio nella chirurgia di questi tumori non deve mai essere minimizzato.
Classifichiamo i tumori come: piccoli, medi, grandi, molto grandi.
I nostri accertamenti indicano che il vostro tumore è:
A) INTRAMEATALE ( ancora dentro l’osso)
B) PICCOLO( inferiore a 1 cm nell’angolo ponto-cerebellare )
C) MEDIO( maggiore di 1 cm, inferiore a 2 cm )
D) GRANDE( maggiore di 2 cm, inferiore a 4 cm )
E) MOLTO GRANDE ( maggiore di 4 cm )
Gestione Dei Neuromi Acustici
Nikolopoulos E O'Donoghue hanno recentemente rivisto 111 articoli sul trattamento del neuroma acustico e ha dichiarato che non esistono metodi "ben programmati per poter effettuare un confronto tra i diversi trattamenti, e quindi la pretesa di medici che sono favorevoli ad un trattamento particolare, sono infondate" (2002). Noi non pensiamo che la saggezza e l’esperienza clinica non debbano essere prese in considerazione , ma certamente al momento attuale ci sembra essere che il trattamento del neuroma acustico sia uno un'arte.
Ci sono tre opzioni distinte:
• gestione medica o attesa vigile "wait and see" [aspetta ed osserva
(trattamento conservativo)]
• intervento chirurgico per rimuovere il tumore
• radioterapia con Gamma-Knife o radioterapia stereotassica
Che linee guida mettere in atto di fronte ad un neurinoma di pochi millimetri nel condotto uditivo interno?
In questi casi ci sono due opzioni che vanno discusse con il paziente. La prima è l’attesa vigile la seconda un intervento chirurgico volto alla preservazione dell’udito. I fattori da considerare nella scelta sono: età e condizioni generali del paziente, udito nell’orecchio interessato e funzione del nervo facciale
E se si tratta di una neoformazione di parecchi centimetri che occupa tutto l’angolo ponto-cerebellare (fase otoneurologica), o peggio con effetto compressivo sul ponte, escavazione del peduncolo cerebellare e del cervelletto (fase francamente neurologica)?
Vanno distinti i tumori in fase otoneurologica da quelli in fase neurologica; quest’ultimi vanno rimossi in toto o in parte al più presto per salvare la vita del paziente mentre per quelli senza segni neurologici esiste la doppia opzione della chirurgia e della radioterapia stereotassica. L’orientamento terapeutico si baserà essenzialmente sulla durata dei sintomi, sulla loro intensità e i desideri del paziente una volta che gli siano stati spiegati i pro e i contro delle due opzioni. Nella tabella che segue sono riportati vantaggi e svantaggi delle due opzioni.
|
|
vantaggi |
svantaggi |
microchirurgia |
asportazione completa diagnosi certa bassissimo rischio per la vita |
bassa percentuale di conservazione dell udito alta percentuale di paralisi facciale postoperatoria |
radioterapia stereotassica con raggi gamma |
nessun rischio chirurgico basso rischio di perdita uditiva basso rischio di paralisi facciale eseguibile in pazienti in cattive condizioni di salute |
permanenza del tumore diagnosi non certa necessità di continui controlli |
Gestione
Assistenza medica per i pazienti con neurofibromatosi di tipo 2 è costituito da esami di routine incentrati sulla diagnosi precoce di alcuni dei potenziali complicazioni legate alla CNS o lesioni del midollo spinale. [2] Gestione da un team di specialisti in una clinica multidisciplinare può fornire la più completa e conveniente cura nel tempo. Quello che segue è uno schema di linee guida ragionevoli nella cura del paziente con NF2:
Esame annuale neurologico cercando deficit sottili o cambiamenti di stato neurologico che potrebbero suggerire la progressione della malattia
Screening uditivo annuale con BAER, con rinvio ad un audiologo per le raccomandazioni di amplificazione, di aumento o di logopedia
MRI annuale per monitorare le lesioni esistenti o per cercare lesioni presintomatica
Valutazioni oftalmologiche annuali per monitorare l'acuità visiva
Gestione Medica (attesa Vigile): Circa il 25% di tutti i neuromi acustici sono trattati con il trattamento medico. L’attesa vigile si attua nei pazienti con neurinoma dell’acustico piccolo nell’unico orecchio udente, nei pazienti che hanno un alto rischio operatorio per altre patologie concomitanti e nei pazienti che rifiutano le altre due modalità di intervento Il trattamento medico consiste nel controllo periodico dello stato neurologico del paziente, l'uso di apparecchi acustici da parte di questi pazienti, se del caso, controlli periodici radiologici(come RM). Si tratta di un appropriato metodo di trattamento per alcuni pazienti (Hoistad et al, 2001). Non esistono farmaci noti che anno un effetto rilevante sulla crescita dei tumori acustici. I tumori possono crescere molto lentamente, a circa 1-1/2 mm per anno, e si può scegliere di seguire un tumore con una serie di esami audiometrici e / o risonanza magnetica (Shin et al, 2000). Nei pazienti di età avanzata, una grave minaccia per la vita o la funzione corporea di crescita tumorale può essere giudicata improbabile nel resto della prospettiva o durata di vita prevista di un paziente e per questo motivo, può essere preferita la gestione medica (Perry et al, 2001). Una volta che un tumore è diagnosticato, la Ia RM viene effettuata dopo 6 mesi e poi con cadenza annuale (Perry et al, 2001).
Questa procedura ha i suoi rischi. Anche quando non si riscontra una crescita del tumore con la RM, vi è sempre il rischio che il paziente perda la funzionalità uditiva utile, rendendo il paziente non più un candidato per la chirurgia conservativa. All’incirca dal 10 al 43% dei pazienti ,seguiti per circa 2 anni, perde quella percentuale di udito “utile” per sentire (Warrick et al, 1999; Shin et al, 2000; Lin et al, 2005).
Trattamento medico sistemico
In uno studio clinico del 2009 presso il Massachusetts General Hospital si è utilizzato il farmaco antitumorale Bevacizumab (nome commerciale: Avastin ) per il trattamento di 10 pazienti con neurofibromatosi di tipo II. Il risultato è stato pubblicato nel New England Journal of Medicine . Dei dieci pazienti trattati con bevacizumab, i tumori si è ridotto a 9 di loro, con il miglior tasso di risposta medio del 26%. La funzione uditiva è migliorata in alcuni dei pazienti, ma i miglioramenti non sono stati fortemente correlati con restringimento del tumore. Bevacizumab agisce tagliando l'apporto di sangue ai tumori e quindi privandoli del loro vettore di crescita. Gli effetti collaterali durante lo studio incluso alanina aminotransferasi , proteinuria e ipertensione (pressione sanguigna elevata) tra gli altri. [8] Uno studio separato, pubblicato in The Neuro-oncologia Journal , mostrano riduzione del tumore del 40% nei due pazienti con NF2, insieme con significativo miglioramento dell'udito Mautner VF, et al. (January 2010).
Nel complesso, i ricercatori credono che il bevacizumab ha mostrato effetti clinicamente significativi sui pazienti NF II. Tuttavia, sono necessarie ulteriori ricerche prima che gli effetti del bevacizumab possAno essere stabilite nei pazienti NF2.
La terapia farmacologica per meningioma NF2 legati
Sunitinib è stato studiato per il trattamento di meningioma che è associato con Neurofibromatosi . [10]
Sono in corso studi per verificare mTORC1 inibitore rapamicina come un potenziale terapeutico in schwannomi NF2 e meningiomi
La terapia farmacologica per schwannoma vestibolare
Lapatinib è stato studiato da Jeffrey Allen presso la NYU Langone Medical Center per il trattamento di schwannoma vestibolare in Neurofibromatosi di tipo II. [11]
L'uso terapeutico di erlotinib ha mostrato risultati promettenti nel trattamento di inoperabili, schwannoma vestibolare progressivo, che comporta non solo una riduzione delle dimensioni del tumore, ma anche nel miglioramento della funzione uditiva. Ulteriori studi clinici sono in ordine prima con questo agente chemioterapico orale può essere consigliato su una base di routine. [16] Una prova di bevacizumab, un fattore di crescita endoteliale anticorpo monoclonale antivascolare, ha anche mostrato una certa efficacia nel ritiro di schwannoma vestibolare; l'udienza farmaco migliorato in alcuni pazienti con tumori non operabili. [Plotkin SR,e 2009]
D'altra parte, non importa quale tipo di trattamento viene utilizzato, nel lungo tempo, è molto improbabile che l'udito rimanga "utile". La cosiddetta "chirurgia conservativa dell’udito " raramente mantiene una funzionalità uditiva utile, questa tende a deteriorarsi abbastanza rapidamente con il tempo, anche se il tumore non è più presente. In base a questi dati , alcuni chirurghi consigliano semplicemente di eliminare tutto l’intero nervo 8°, in quanto questa soluzione rende molto improbabile una recidiva del tumore. A nostro parere, questo è un giudizio opportuno - ma non un'idea irragionevole. Una stima ragionevole è che dopo più di un anno, circa il 75% dei tumori avrà un allargamento visibile, una media di 1,5 millimetri, e circa il 25% non lo avrà. Alcune varietà crescono molto più rapidamente di altri.
Nei pazienti con la neurofibromatosi, l'udito è destinato a rimanere stabile nel orecchio operato per circa 1-2 anni (Masuda et al, 2004).
La Radioterapia con raggi gamma ha la finalità di bloccare l’accrescimento del tumore e ottiene questo in un’alta percentuale di casi con minori rischi e complicanze della chirurgia. Naturalmente il tumore rimane e deve essere controllato nel tempo.
Gamma Knife: quando il rischio di intervento chirurgico è elevato a causa di altri problemi di salute, o se il paziente rifiuta la chirurgia, può essere utilizzata la "gamma Knife " . Questo è un metodo di irradiare il tumore, inventato da Lars Leksell nel 1971. Questa procedura evita un intervento chirurgico con i rischi che ne possono derivare. In passato, questa opzione è stata generalmente consigliata solo per i casi , a più elevato rischio chirurgico, a causa della possibilità di complicazioni tardive da radiazione, e la necessità di un monitoraggio continuo con RM dei risultati della procedura.
Non si consiglia utilizzare la gamma knife ad alte dosi a causa della possibilità di complicazioni da radiazione dopo e oltre i 2 anni. Tuttavia, gamma knife a basso dosaggio è alla ricerca molto meglio e ci sono sicuramente molte volte quando è l'opzione migliore. Basse dosi di radiazioni (ad esempio 13 Gy) sono attualmente consigliato a causa del rischio molto più basso di debolezza e intorpidimento facciale (Wackym et al, 2004).
Una conseguenza interessante del protocollo di radiazioni a basso dosaggio è che si è riscontrato che i pazienti, dopo le radiazioni, non hanno una perdita completa dell'udito o della funzione vestibolare. In alcuni casi questa ridotta funzionalità vestibolare può essere molto fastidiosa in quanto può provocare sintomi di irritabilità nervosa, come ad esempio l'iperventilazione che può indurre del nistagmo. Ciò è una probabile conseguenza di utilizzare una metodologia di trattamento che agisce più lentamente rispetto ad un intervento chirurgico. Sembra probabile che questa complicanze siano più comune nelle pazienti che hanno tumori di piccole dimensioni.
In queste situazioni l'iperventilazione induce un nistagmo che batte verso la lesione (a differenza del nistagmo provocato dalle vibrazioni che batte in direzione opposta alla lesione). Spesso è un nistagmo molto intenso . Nei pazienti con questo segno, ci si può aspettare che scompaia (questa scomparsa del nistagmo indotto da iperventilazione potrebbe richiedere diversi anni), si può provare qualche farmaco che riduca l'irritabilità del nervo, in caso contrario si riconsiderare il trattamento chirurgico.
 Materiale supplementare sul sito DVD : Video di nistagmo indotto da iperventilazione in pazienti con neuroma acustico sul lato sinistro
Materiale supplementare sul sito DVD : Video di nistagmo indotto da iperventilazione in pazienti con neuroma acustico sul lato sinistro
Radioterapia stereotassica. Radiazione diversi dai raggi gamma possono essere utilizzati anche per il trattamento di neuroma acustico. Essi comprendono l’acceleratore lineare (LINAC) e Cyberknife. Le altre modalità sono simili a Gamma Knife nelle caratteristiche generali. La conservazione dell'udito a lungo termine è molto raro in persone con radioterapia stereotassica (6,7% secondo Lin et al, 2005). In altre parole, anche se l '"obiettivo" è quello di preservare l'udito, praticamente questo non è realistico. Non vediamo alcun motivo particolare per utilizzare la radioterapia stereotassica, piuttosto che Gamma Knife. La probabilità di recidiva del tumore utilizzando regimi di dosaggio attuale è di circa il 5-10%. La crescita del tumore è rara in pazienti che rimangono stabili dopo 6-7 anni di terapia .
I Problemi della radioterapia sono le recidive (5-10%), perdita dell’ udito (alla fine il 93%), il rischio di irradiare tumori di grandi dimensioni (> 2 cm) dovute al rigonfiamento del tumore nel primo anno, il rischio di malignità (Tanbouzi et al, 2011 , Markou et al, 2011), idrocefalo (raro), aneurisma rotto IAC (raro), e accelerata aterosclerosi vertebro-basilare (es. Jackler, 2007).
Gestione di perdita di udito in NF2 Impianti uditivi del tronco encefalico
Perché la perdita dell'udito in quelli con NF2 si verifica quasi sempre dopo l'acquisizione di competenze linguistiche verbali, i pazienti non si integrano sempre bene nella cultura dei sordi e sono più propensi a ricorrere a uditivo tecnologie assistive . Il più sofisticato di questi dispositivi è l' impianto cocleare , che a volte può ripristinare un alto livello di funzione uditiva anche quando udito naturale è totalmente persa Schwartz MS,e 2008. . Tuttavia, la quantità di distruzione al nervo cocleare causato dalla tipica schwannoma NF2 spesso preclude l'utilizzo di tale impianto. In questi casi, un impianto uditivi del tronco encefalico (ABI) in grado di ripristinare un livello primitivo di udito, che, quando è integrato da lettura labiale , in grado di ripristinare una conoscenza funzionale della lingua parlata.
|
|
Fig.-9 Neuroma acustico molto grande (proiezione coronale, il tumore è la grande macchia bianca). Fonte: Mayo Clinic Neuroscience Update. |

Trattamento chirurgico: Lo scopo primario della chirurgia del neurinoma dell'acustico è l'exeresi totale del tumore conservando, ove possibile, l'integrità anatomica e funzionale dei nervi cranici, con le minori sequele circa la metà di tutti i neuromi acustici sono attualmente trattati con la chirurgia. Questa opzione probabilmente diminuirà nei prossimi decenni, per l'uso crescente del trattamento con gamma knife . La figura sopra mostra un neuroma acustico ,di grandi dimensioni ,in questi casi è da preferire il trattamento chirurgico. Nella maggior parte dei casi la rimozione chirurgica del tumore è l'opzione preferita perché previene complicanze potenzialmente fatali di crescita del tumore (anche se questa evenienza è piuttosto insolita).L’intervento chirurgico potrebbe in teoria permettere di "conservare" la funzionalità uditiva, anche se è molto raro che l'udito sia effettivamente utile dopo l'intervento chirurgico. Di solito l'intervento viene effettuato presso un centro universitario da un team di chirurghi compreso un neurotologo (un otorinolaringoiatra specializzato) e un neurochirurgo. Ci sono diversi approci operativi.
|
|
Fig.-10 a Comuni approcci chirurgici per il Neuroma Acustico testa. Translabirintica (attraverso l'orecchio interno). La perdita dell'udito è previsto e inevitabile. Non adatto per i tumori di grandi dimensioni. Retrosigmoidea o suboccipitale (attraverso il craniodietro l'orecchio). Retrazione del cervelletto (parte del cervello) è necessaria. Mal di testa sono comuni dopo tale approccio. (Immagine da Jackler R, Atlas of Neurotology e Skull Base Surgery (Mosby 1996, prima edizione). |

Ciascuno di questi approcci chirurgici ha vantaggi e svantaggi che devono essere considerati nella scelta di un approccio ottimale. Il trattamento chirurgico in cui il cervello è esposto è quasi sempre eseguita da un team di chirurghi, di solito sono presenti un neurootologo ed un neurochirurgo. La maggior parte dei pazienti vengono ricoverati in ospedale il giorno prima dell'operazione. Dopo l'intervento chirurgico, è bene trascorrere la 1a notte in una unità intensiva. La maggior parte dei pazienti sono dimessi dall'ospedale entro 4-6 giorni dal'intervento chirurgico, ed entro 6 settimane di solito è possibile riprendere l'attività lavorativa .La RM di solito viene effettuata dopo 1 e 5 anni per rilevare eventuali residui o recidive tumorali. Si consiglia la rimozione del tumore totale o quasi totale (95%) (Sanna et al, 2002). Si consiglia un attento follow-up con ricorrenti RM .La presenza di noduli od un allargamento progressivo del condotto uditivo interno può rappresentare una ricrescita del tumore (Brors et al, 2003).
I tumori che si estendono nel labirinto stesso (vale a dire "gli schwannomi intracocleare") non possono essere rimossi con l'approccio retrolabintico " e vi possono essere recidive ai tumori, e possono ripresentarsi. Ci possono essere molte complicazioni importanti, che possono derivare da un intervento chirurgico , queste complicazioni debbono essere prese in considerazione (vedi sotto).
riguardo alle chirurgia conservativa dell'udito , conservazione sempre auspicabile, purtroppo, la possibilità che l'udito sia preservato dopo un intervento chirurgico di neuroma acustico è minimo. Nedzelski e colleghi hanno recentemente riferito che solo il 16% circa dei pazienti ha avuto un udito utilizzabile nei follow-up dopo l'intervallo di "chirurgia conservativa dell'udito". (Lin et al, 2005). Mentre la funzionalità uditiva può essere "conservava" immediatamente dopo l'intervento chirurgico, di solito si deteriora in pochi anni.
Prognosi
La prognosi di neurofibromatosi tipo 2 (NF2) dipende da una serie di fattori, tra cui l'età di insorgenza dei sintomi, il grado di sentire deficit, e il numero e la posizione dei vari tumori. Anche se l'età di insorgenza è relativamente simile all'interno delle famiglie, la fascia d'età può variare 2-70 anni. Mentre i tumori stessi sono relativamente indolente e non subiscono la trasformazione maligna, studi effettuati alla fine del 1980 e all'inizio del 1990 hanno mostrato chiaramente che i tassi significativi di mortalità e morbilità sono associati con la diagnosi di NF2.
Uno di questi studi ha suggerito che la sopravvivenza dal momento della diagnosi medie effettive a 15 anni; tuttavia, questo potrebbe cambiare in meglio con diagnosi migliore, tecniche chirurgiche, la sorveglianza, lo screening e il riconoscimento di malattia lieve (dovuto in parte alla maggiore consapevolezza del medico e la disponibilità di opzioni di diagnostica molecolare).
Morbilità e mortalità
Schwannoma vestibolare sono la caratteristica più comune e ben riconosciuto di NF2 che porta a una significativa morbidità. I sintomi di tinnito, perdita dell'udito progressiva, e anche la disfunzione vestibolare sono spesso i primi segnali di NF2.Anche se la perdita uditiva unilaterale è il numero 1 che presenta sintomi, sordità bilaterale sarebbe previsto per eventualmente verificarsi in individui più colpiti.Schwannoma vestibolare non trattati possono estendere a livello locale e possono causare compressione del tronco cerebrale, idrocefalo, e, occasionalmente, paralisi del nervo facciale.
Schwannomas midollo spinale a forma di manubrio sono abbastanza comuni in NF2 e determinano una significativa morbilità; essi presentano una grande sfida terapeutica. Ependimoma midollo spinale, astrocitomi e meningiomi si verificano anche, ma meno frequentemente. Meningiomi intracranici, d'altra parte, è frequente; essi possono essere asintomatici, o possono causare una varietà di sintomi e deficit del SNC 10. Aboukais R,e 2010.
Schwannomas Nonvestibular si verificano in più della metà dei pazienti e sono spesso diagnosticati in pazienti con una precedente età alla diagnosi di NF2.Nervi cranici III e V sono più comunemente coinvolti, ma la rara presenza di schwannomas forame giugulare potenzialmente impattare il glossofaringeo, vago, e / o nervi spinali accessorio può portare a disfagia, disturbi della motilità esofagea, raucedine, o aspirazione.
D'altra parte, schwannoma nonvestibular in pazienti con NF2 tendono ad essere più indolente e crescere lentamente nel tempo. Questo può complicare il trattamento del processo decisionale, dal momento che le opzioni includono la chirurgia, la radioterapia e vigile attesa Fisher LM,e 2008.
Subcapsulare posteriore, o giovanile, la cataratta può precedere CNS sintomatologia. Le cataratte possono progredire nel corso del tempo, portando a riduzione dell'acuità visiva. Una buona percentuale di individui affetti si trovano ad avere amartomi retinici o membrane epiretiniche che possono o non possono essere visivamente notevole.
Polineuropatia sensoriale motore è visto in alcuni individui con NF2 che possono o non possono avere tumori identificabili lungo la lunghezza del nervo periferico (s) di interesse.
Educazione del paziente
I pazienti ed i membri a rischio della famiglia dovrebbero essere resi consapevoli dei sintomi specifici, come tinnito, deficit dell'udito, debolezza focale, cambiamenti sensoriali, o problemi di equilibrio, che potrebbe suggerire la crescita del tumore e deve richiedere l'attenzione medica immediata.
I pazienti con schwannoma vestibolare devono essere avvertiti circa le immersioni e attività subacquee, a causa di un aumentato rischio di disorientamento e il potenziale di annegamento.
I pazienti e le loro famiglie possono essere attribuiti alla neurofibromatosi (NF)-specifici gruppi regionali e nazionali di sostegno per continui aggiornamenti sui progressi nel trattamento, nonché per il sostegno emotivo. Neurofibromatosi, Inc., per esempio, può essere raggiunto al numero verde 1-800-942-6825.
La NF2 Review, che si trova a Los Angeles, CA, può essere raggiunto a 1-213-483-4431 e non solo ha un bollettino specifico NF2, ma ha anche un particolare interesse di ricerca in NF2, con un team di specialisti che lavorano su uditiva Tronco cerebrale impianti (Abis) e le più recenti approcci chirurgici per schwannoma vestibolare.
Risorse online includono il sito Web NIH e di un gruppo di sostegno NF2 da persona a persona conosciuta come l' equipaggio NF2 .
Approccio dell Dr. Hain's.
Si suggerisce ai potenziali candidati per un intervento chirurgico in primo luogo considerare la sicurezza e la probabilità di complicazioni quando si deve prendere in considerazione la chirurgia o gamma knife. Se uno ha un udito utilizzabile , e non c'è altro pericolo di aspettare (come ad esempio se necessitano di una operazione più importante ), si potrebbe ragionevolmente semplicemente aspettare fino a che la funzionalità uditiva diventi inutilizzabile, prima di procedere con la chirurgia o radioterapia. In questi casi la procedura dovrebbe essere : controlli periodici della funzionalità uditiva e meno frequentemente RM . La frequenza delle prove è determinato principalmente dal tasso di variazione nelle misure, ma circa ogni 6 mesi per l'esame audiometrico e circa una volta / anno per l'esame MRI e di solito una frequenza appropriata
Sia un intervento chirurgico che e gamma-knife sembrano dei metodi ragionevoli per la gestione dei tumori acustica , la scelta dipende da fattori individuali. La gestione chirurgica ha il vantaggio che si "ottengono dei risultati con rapidità ". La gamma knife è meno stressante all'inizio , ma può ritardare la risoluzione delle vertigini.
Riabilitazione Vestibolare
Nella maggior parte dei casi, i risultati della chirurgia del neuroma acustico comportano una perdita completa della funzione vestibolare sul lato operato. La Gamma knife inoltre è associato ad una diminuzione sostanziale della funzione vestibolare (Wackym, 2004). A seconda della quantità della funzione vestibolare presente prima di un intervento chirurgico, i pazienti possono avere vertigini o disturbi dell'equilibrio dopo l'intervento (Levo et al, 2004). La riabilitazione vestibolare può accelerare il recupero di questo deficit. A meno che non vi sia già una completa perdita della funzione vestibolare prima dell'intervento chirurgico o delle radiazioni (come documentato con la ENG), pensiamo che sia meglio che il paziente che intende sottoporsi ad un'operazione chirurgica per neuroma dell’ acustico visiti di un fisioterapista vestibolare per assicurarsi che sia in " buona forma ", e che imparare le procedure di base, e per iniziare un programma singolo settimanale di PT per 1-2 mesi dopo la dimissione. È importante che il chirurgo otologo che esegue l'operazione sia coinvolto con la terapia, in qualche modo (ad esempio nei pazienti con perdita di CSF), in questi casi la terapia deve essere ritardata.
Popolazioni speciali e trattamento
Piazza e altri di recente hanno suggerito che negli anziani, dovrebbe essere seguito il presente algoritmo: Se il neurinoma acustico protrude meno di un 1 cm i nell'angolo ponto-cerebellare, una risonanza magnetica dovrebbe essere ripetuta entro in un anno. Se il tasso di crescita è <2 mm / anno, il paziente deve essere osservato. Se la velocità è superiore superiore , bisognerebbe proporre la chirurgia .
Secondo Piazza, per i tumori che> sporgono 1 centimetro nel’ angolo PC (ponto-cerebellare), ai pazienti in buona salute generale, dovrebbe essere proposto un intervento chirurgico. Nei pazienti in cattive condizioni di salute generale, dovrebbe essere proposta la radiochirurgia (Piazza et al, 2003)
Complicanze della Chirurgia
Il trattamento chirurgico, di per sé, ha un rischio notevole. Nel complesso, il rischio di
morte dovuto ad un intervento chirurgico di neuroma acustico è di circa 0,5 al 2 per cento. Complicanze post-operatorio Impreviste si verifica in circa il 20 per cento , ancora più complicazioni si verificano nei soggetti anziani e infermi e quelli con tumori di grandi dimensioni (Kaylie et al, 2001). Complicazioni, ordinato da rare a frequenti, sono elencati.
• Stroke (raro)
• Lesioni al cervelletto, ponte o del lobo temporale (rare)
• morte (all in circa 0,5 per cento al 2%, a seconda del centro)
• perdita di CSF (5-15 per cento di tutti i pazienti)
• Meningite (2-10 per cento)
• Debolezza facciale (incidenza dal 4 al 15 per cento di paralisi totale). Le dimensioni del tumore è un grande fattore di rischio per questa complicanza.
• rischio di perdita dell'udito (immediatamente dopo l'intervento chirurgico)
•Translabirintica - 100%
•retrosigmoidea - 60%
• Fossa cranica Media --40%. L'udito occasionalmente migliora con l'approccio attraverso la fossa media (Stidham e Roberson, 2001).
• Mal di testa (persistenti nel 10-34 per cento, Ruckenstein et al 1996; Soumekh et al, 1996)
• Squilibrio e vertigini (quasi in tutti i pazienti che hanno una funzione vestibolare prima dell'intervento, avranno un peggioramento dell'equilibrio dopo l'intervento chirurgico).
APPROFONDIMENTO DEI RISCHI E COMPLICAZIONI DELLA CHIRURGIA DEI TUMORI DEL NERVO ACUSTICO
Non è possibile elencare in modo dettagliato tutte le complicanze che possono intervenire prima, durante e dopo l’atto chirurgico.
Il seguente elenco è stato realizzato allo scopo di indicare alcuni dei principali e più importanti rischi possibili della chirurgia dei tumori del nervo acustico.
1 - PERDITA DELL’UDITO
Nei piccoli tumori è talora possibile salvare l’udito rimuovendo il tumore ( vedi paragrafo sui problemi relativi alla conservazione dell’udito ).
La maggioranza delle neoplasie sono comunque grandi e l’udito dell’orecchio interessato viene perso in seguito alla rimozione chirurgica. Il paziente potrà continuare una vita del tutto normale con un solo orecchio udente.
2 – RUMORE (acufeni)
Nella maggior parte dei casi il rumore rimane inalterato, come prima dell’intervento. Nel 10% dei casi dopo l’intervento può essere più intenso, in alcuni casi ( 20-30 % circa ) il rumore può anche scomparire.
3 - DISTURBI DEL GUSTO E SECCHEZZA DELLA BOCCA
I disturbi del gusto e la secchezza della bocca sono comuni, per alcune settimane, dopo l’intervento. Nel 5% dei pazienti questi disturbi possono durare più a lungo.
4 - VERTIGINE E DISTURBI DELL’EQUILIBRIO
Nella chirurgia dei tumori del nervo acustico è necessario anche rimuovere una parte o tutto il nervo dell’equilibrio. Nella maggior parte dei casi è, inoltre, necessario rimuovere il labirinto.
Poiché il nervo dell’equilibrio è spesso danneggiato dal tumore, la sua rimozione quasi sempre comporta un miglioramento dell’instabilità preoperatoria.
Tuttavia la vertigine è comune nel post operatorio e può essere anche grave per alcuni giorni o per qualche settimana. Disequilibrio, o instabilità con il movimento della testa, possono essere presenti per lungo tempo ( nel 30% dei pazienti ), fino a che non avviene il compenso da parte degli altri centri dell’equilibrio.
5 - PARALISI DEL NERVO FACCIALE
I tumori del nervo acustico sono in intimo contatto con il nervo facciale ( nervo che determina il movimento sia dei muscoli deputati alla chiusura dell’occhio, sia quelli che controllano l’espressione del viso ). In seguito all’asportazione di un tumore del nervo acustico può verificarsi una paralisi temporanea dell’emiviso interessato, ciò comporta un’asimmetria del volto con impossibilità a chiudere l’occhio.
La paralisi del nervo facciale può essere la conseguenza di un edema, di una lesione diretta del nervo o di una compromissione della vascolarizzazione .
Solo tramite un’attenta chirurgia con l’ausilio del microscopio e del monitoraggio intraoperatorio del nervo facciale è possibile conservare l’integrità e la funzionalità del nervo. Nonostante ciò, una paralisi transitoria del facciale può insorgere ugualmente.
Generalmente il recupero avviene spontaneamente.
Il recupero della funzionalità facciale, è molto variabile da soggetto a soggetto; può avvenire nel giro di alcune settimane o di alcuni mesi (fino a 10-12 mesi) e può non essere completa al 100%. Oltre questo termine le probabilità di recupero sono molto ridotte ed il chirurgo può proporre al paziente l’esecuzione di un secondo intervento che ha lo scopo di collegare il nervo facciale all’undicesimo nervo cranico ( anastomosi ipoglosso-facciale ) onde evitare l’atrofia dei muscoli della faccia e permettere la chiusura dell’occhio.
Nel 5 % dei casi il nervo facciale è completamente avvolto o inglobato dalla massa tumorale. In casi molto rari il tumore può anche originare direttamente dal nervo facciale ( neurinoma del nervo facciale). In entrambe queste condizioni, è necessario interrompere il nervo facciale per poter eseguire l’asportazione del tumore. In tal caso può essere possibile ricostruire immediatamente il nervo mediante un trapianto. Se ciò non è possibile, si può effettuare in un secondo tempo un intervento per collegare il nervo facciale ad un nervo del collo ( anastomosi ipoglosso-facciale). Debolezza facciale di vario grado , ma la debolezza grave con i punteggi di House- Brackman V-VI A ad 1 anno si sono verificati nel 6% (Mass et al, 1998). Wiet e altri hanno recentemente riportato risultati su 500 casi (Wiet et al, 2001).
6 - COMPLICAZIONI OCULARI
La paralisi del nervo facciale spesso rende l’occhio asciutto e privo di difese. La consulenza di un oculista può essere indicata. Talora si rende necessario applicare delle lacrime artificiali, utilizzare un occhialino protettivo e/o un pesino palpebrale esterno o parziale chiusura chirurgica delle palpebre per ridurre il rischio di una cheratite o di un’ulcera corneale. Questi accorgimenti mantengono l’occhio umido evitando così le complicazioni oculari.
7 - DIMINUZIONE DELLA FUNZIONALITÀ DI ALTRI NERVI NEI GRANDI TUMORI
Il tumore del nervo acustico può prendere contatto con i nervi che controllano i muscoli oculari, della faccia, della bocca e della gola. Questi nervi possono essere lesi e provocare una visione doppia, intorpidimento della gola, della faccia e della lingua, debolezza della motilità della spalla, disfonia e difficoltà alla deglutizione.
Questi problemi possono rimanere permanentemente. Tale eventualità è però molto rara.
8 - COMPLICAZIONI CEREBRALI E MORTE
I tumori del nervo acustico sono localizzati vicino ai centri cerebrali vitali che controllano la respirazione, la pressione sanguigna e la funzionalità cardiaca. Poiché il tumore ha una crescita progressiva, può venire a contatto con questi centri cerebrali e può inglobare vasi sanguigni che irrorano queste aree del cervello. Un’ attenta e scrupolosa dissezione del tumore permette, generalmente, di evitare tali complicanze. Se l’irrorazione sanguigna di questi centri cerebrali è compromessa, possono instaurarsi serie complicanze: perdita del controllo muscolare, paralisi ed anche la morte. Vi è una incidenza complessiva dello 0,5 per cento di morte a causa di un intervento chirurgico neuroma acustico .Nella esperienza del gruppo si SANNA, la mortalità peri-operatoria è inferiore all’ 1% nei piccoli tumori e medi; tale percentuale aumenta nei tumori grandi o giganti ( 2-3 % ).
9 - PERDITA POSTOPERATORIA DI LIQUIDO CEREBROSPINALE MENINGITE
La chirurgia dei tumori del nervo acustico comporta una perdita temporanea di liquido cerebrospinale ( il liquido che avvolge il cervello ). Per evitare tale complicanza dopo la rimozione del tumore, la cavità viene riempita con grasso prelevato dall’addome. Occasionalmente la fuoriuscita di liquido può presentarsi dopo l’intervento e per farla cessare può essere necessario reintervenire oppure inserire un drenaggio lombare.
Il controllo e la risoluzione immediata della fuoriuscita di liquor è determinante per evitare l’insorgenza di una meningite, che è una temibile infezione del liquido e del tessuto attorno al cervello. Quando si ha questa complicanza, è necessaria un’ospedalizzazione prolungata.
E’ spesso indicato inoltre un trattamento con elevati dosaggi di antibiotici. Una recente revisione delle complicazioni dell’ intervento chirurgico ha riscontrato che le complicanze più comuni sono perdite di CSF (9,4%) e la meningite (1,5%) (Slattery et al, 2001).
10 - EMORRAGIA POSTOPERATORIA ED EDEMA CEREBRALE
Emorragia ed edema cerebrale possono svilupparsi dopo la chirurgia per tumore del nervo acustico (1%) . Se accade, può essere necessario un ulteriore intervento, d’urgenza, per aprire la ferita, bloccare il sanguinamento e permettere al cervello di riespandersi.
L’emorragia è una complicazione molto seria, e rappresenta un’urgenza chirurgica per cui il paziente viene sottoposto a continui monitoraggi in terapia intensiva.
12 - REAZIONI DA TRASFUSIONE DI SANGUE
Raramente è necessario praticare delle trasfusioni di sangue durante un intervento per tumore del nervo acustico. Reazioni immediate sono rare. Una complicanza tardiva della trasfusione è un’ infezione virale del fegato ( epatite); evento molto raro in quanto la legge obbliga i donatori ad esami specifici prima della donazione.
il
successo complessivo di conservare l una funzionalità uditiva era del
27%, i risultati nettamente migliori si sono ottenuti durante quando il paziente veniva operato attraverso la fossa media .
Anche se non proprio una complicazione, l'udito all'orecchio operato spesso si deteriora nel tempo in misura maggiore di quanto l'orecchio non operato, anche in assenza di recidiva neoplastica (Chee et al. 2003). La percentuale di persone con "udito utilizzabile" può deteriorarsi del 25% nel tardo post-operatorio. Questo peggioramento uditivo è stata variamente attribuita: alla cicatrizzazione, fibrosi, o microemorragie durante l'intervento . Nel lungo periodo, pochissimi pazienti conservano una funzionalità uditiva utile (Lin et al, 2005). Alcuni autori hanno suggerito che, dato la modesta funzionalità uditiva che viene recuperato in pochi pazienti che sono candidati ad un intervento chirurgico per preservare l'udito risparmiatori di, che la conservazione dell'udito non si ritiene utile una chirurgia del neuroma acustico che abbia come ’obiettivo per la conservazione dell'udito (Tos et al, 1988). La nostra opinione è che di tanto in tanto vale la pena tentare, ma le aspettative devono essere realistiche.
L'equilibrio peggiora dopo l'intervento del neuroma acustico a causa dei danni chirurgia o per la rimozione del nervo vestibolare (Levo et al, 2004). A lungo termine (cioè 2 anni), l la funzionalità vestibolare torna in generale a quasi normale, ma i pazienti che hanno deficit sensoriali i, come ad esempio disturbi visivi (cataratta ), perdita di sensibilità (neuropatia ), danni al cervello (ad danni cerebellari o al tronco cerebrale causati dal tumore o dal’intervento chirurgico), o scarso adattamento (ad esempio a causa dell'età avanzata) non può mai ottenere la restituzione completa dell’ equilibrio.
In una recente riunione presso l'ANA (Associazione del neuroma acustico) si è partecipato ad una sessione di 2 ore e intervistato 8 pazienti che avevano vertigini persistenti. Come regola generale, i pazienti che avevano vertigini 2 anni dopo l'intervento, avevano avuto contemporaneamente una lesione del cervelletto ed una totale perdita vestibolare, associata ad un difficile intervento chirurgico (di solito per un tumore > 4 centimetri). La risonanza magnetica di uno di questi pazienti è i sotto riportata
|
|
|
Fig.-22 RM di paziente che aveva un neuroma acustico rimosso per mezzo di approccio retro labirintico e con vertigini refrattarie i. Sul lato destro della foto (a sinistra della testa), si trova una zona nera dove c'era un danneggiamento del cervelletto, presumibilmente connessi con la chirurgia. |

Nella pratica clinica dell'autore a Chicago Illinois , si suggerisce che i potenziali candidati alla chirurgia operativi considerano prioritaria la sicurezza e la probabilità di complicazioni quando si considera la chirurgia o Gamma Knife. Se uno ha un udito utile, e non c'è altro pericolo di attesa (ad esempio, necessitano di un funzionamento più grande), ci si potrebbe ragionevolmente attendere fino a quando l’udito diventa inservibile prima di procedere con la chirurgia o la radioterapia. Ecco la procedura sarebbe: periodici test dell'udito e le scansioni RM meno frequenti. La frequenza delle prove è determinata principalmente dal tasso di variazione nelle misure, ma circa ogni 6 mesi test audiometrici e circa una volta l’anno scansione RM sono in genere appropriati.
Entrambi i metodi di chirurgia e di gamma-knife di gestione tumori acustici sembrano ragionevoli a questa scrittura, con la scelta dipende da fattori individuali. Il trattamento chirurgico ha il vantaggio che essa "viene finita in fretta". Gamma Knife è meno stressante acuto, ma può ritardare la risoluzione di vertigini.
Riabilitazione vestibolare
Nella maggior parte dei casi, come risultato della chirurgia del neurinoma dell’acustico si ha una perdita completa della funzione vestibolare sul lato operato. Anche il trattamento con Gamma Knife è associato ad un un sostanziale declino della funzione vestibolare (Wackym, 2004). I pazienti spesso sperimentano vertigini e disequilibrio post-chirurgico (Levo et al, 2004; Tufarelli et al, 2007). La riabilitazione vestibolare può accelerare il recupero da questo deficit. A meno che non ci sia già una perdita completa della funzione vestibolare prima dell’ intervento chirurgico o della radioterapia (come documentato in ENG ), pensiamo che sia meglio che il paziente che sta progettando di essere sottoposto ad un intervento chirurgico di neurinoma dell’acustico consulti un fisioterapista vestibolare per fare in modo che ci sia una " buon adattamento "e impari le procedure di base, e per l'individuo di iniziare un programma settimanale di riabilitazione per 1-2 mesi dopo la dimissione. È importante che l’otochirurgo che comunque esegue parte della terapia come in alcune situazioni (ad esempio pazienti con perdita del CSF ), in questi casi la terapia riabilitativa deve essere ritardata.
Gestione di popolazioni speciali
Piazza e altri recentemente suggeriscono che negli anziani, il seguente algoritmo dovrebbe essere seguito: Se il neurinoma dell’acustico sporge meno di 1 cm in ponto-cerebellare, una risonanza magnetica deve essere ripetuto in un anno. Se il tasso di crescita è <2 mm / anno, occorre osservare il paziente. Se valori maggiori di quelli soprariportati , offerto chirurgia.
Secondo Piazza, per i tumori che sporgono> 1 cm nel angolo CP, pazienti in buona salute generale dovrebbero essere offerti chirurgia. Ai pazienti in cattive condizioni di salute generale, dovrebbero essere offerta la radiochirurgia (Piazza et al, 2003).
Le complicazioni e le conseguenze di un intervento chirurgico per neurinoma acustico:
Il trattamento chirurgico, di per sé, ha un rischio sostanziale. Complessivamente, il rischio di morte dalla chirurgia neuroma acustico è di circa 0,5 a 2 per cento . Inaspettate complicanze post-operatorie si verificano in circa il 20 per cento con più complicazioni che si verificano in individui anziani e infermi e quelli con tumori di grandi dimensioni (Kaylie et al, 2001).
Le complicazioni, ordinate da rare a frequenti, sono sottoelencati.
Corsa (raro)
Lesioni al cervelletto, ponte o lobo temporale (raro oggi (2013), ma più comune in passato)
Decessi (circa 1/2 per cento al 2%, a seconda del centro)
Perdita di CSF (5-15 per cento di tutti i pazienti)
Meningite (2-10 per cento)
Paralisi del facciale (4 al 15 per cento l'incidenza di paralisi totale). La dimensione del tumore è un grande fattore di questa complicanza.
Sentendo il rischio di perdita (subito dopo l'intervento chirurgico)
Translabirintica - 100%
Retrosigmoidea - 60%
Fossa media - 40%. La funzionalità uditiva occasionalmente migliora per l'approccio della fossa media (Stidham e Roberson, 2001).
Mal di testa (persistente nel 10-34 per cento, Ruckenstein et al 1996; Soumekh et al, 1996)
Disequilibrio e vertigini (quasi tutti i pazienti che hanno una funzione vestibolare prima dell'intervento, si ha un peggioramento dell’equilibrio dopo l'intervento chirurgico). Questa non è una complicazione, ma una conseguenza.
Una recente revisione delle complicazioni di un intervento chirurgico suggerisce che le perdite del QCS (9,4%) e la meningite (1,5%) sono le complicanze più comuni (Slattery et al, 2001).
Lesione cerebellare:
|
|
|
|
Risonanza magnetica da persona che ha avuto neuroma acustico rimosso tramite approccio retrolab, e con vertigini refrattario. Sul lato destro della figura (lato sinistro della testa), c'è una zona nera dove erano presenti danni al cervelletto, presumibilmente associati alla chirurgia |
Un'altra scansione che mostra i danni cerebellare dopo chirurgia neuroma acustico. Questo tipo di lesione sarà probabilmente accompagnata da squilibrio permanant grazie alla combinazione di una lesione cerebellare e perdita di funzione dell'orecchio interno sulla sinistra. |
|
|
|
|
Tumore operato nel 1961. Vi è un chiaro danno cerebellare sul lato sinistro del cervelletto (lato destro della figura). |
Neuroma acustico residuo nonché danni cerebellare, in questo paziente che ha avuto un intervento chirurgico nel remoto passato. |




In una revisione dei risultati di 258 pazienti operati con approccio translabirintica, ictus o lesioni del cervelletto cerebellar injury si sono verificato nel 1,1%. Lesione cerebellare può verificarsi a causa di trazione, nonché a causa di lesioni alla arteria cerebellare inferiore anteriore (Hegarty et al, 2002). Immagini di lesione cerebellare da trazione sono sopra riportate . Questi pazienti hanno generalmente segni oculomotori molto importanti (ad esempio, nistagmo) e disequilibrio persistente (ad esempio per tutta la vita).
Decessi
Vi è nel complesso una incidenza dello 0,5 per cento di morte a causa di un intervento chirurgico per neurinoma dell’acustico. Vi è anche le possibilità di assistenza per la riabilitazione a lungo termine (1,2%)ed a breve termine (4,4%) (Barker et al, 2003). Pertanto, vi è più o meno una possibilità dell 1,7% di morte o di assistenza o cura necessaria a lungo termine dopo l'intervento chirurgico di neuroma dell’acustico. Le probabilità sono migliori se si sceglie un ospedale e chirurgo con"alto volume" di interventi.
Altre complicazioni includono perdita del CSF nel 7,8%, la meningite a 1,6%.
Paralisi facciale
Paralisi facciale di vario grado apparso in più, ma una grave paralisi del facciale, di grado V-VI con la classificazione di House-Brackman ad 1 anno si è verificato nel 6% (Messa et al, 1998). Wiet e altri hanno recentemente riportato risultati in 500 casi (Wiet et al, 2001). Successo complessivo a mantenere una funzionalità uditiva utile è stata del 27%, con risultati decisamente migliori ottenuti tramite l'approccio della fossa media.
la conservazione dell’udito con l'intervento chirurgico è di solito solo un pio desiderio.
l’udito all'orecchio operato spesso si deteriora nel tempo in misura maggiore dell'orecchio non operato, anche senza un tumore recidivante (Chee et al. 2003). La percentuale di persone con "udito funzionale" potrebbe deteriorarsi del 25% nel periodo post-operatorio . Questo è stato variamente attribuito a cicatrici, fibrosi, o micro emorragie durante l’operazione.
Nel lungo termine, pochissimi pazienti conservano un udito utilizzabile (Lin et al, 2005). Alcuni autori hanno suggerito che, dato la modesta funzione uditiva che è recuperata in pochissimi pazienti che sono candidati per sentire con la chirurgia conservativa, la conservazione dell'udito come obiettivo della chirurgia neuroma acustico non è utile (Tos et al, 1988). La nostra opinione è che di tanto in tanto la pena di tentare, ma le aspettative di uno devono essere realistico.
L'equilibrio generalmente peggiora dopo l'intervento chirurgico neuroma acustico
L'equilibrio si deteriora dopo l'intervento del neurinoma acustico perché la chirurgia danneggia o rimuove la rimantenente funzione del nervo vestibolare (Levo et al, 2004; Tufarelli et al, 2007). Tufarelli e associati hanno riferito che il 10% dei 459 pazienti ha giudicato il loro squilibrio come invalidante, e il 73% ritiene di avere una oscillopsia moderata (difficoltà a vedere con la testa in movimento)
E 'stata la nostra esperienza che nel lungo periodo (cioè 2 anni), bilancia ritorna in generale a quasi normale, ma le persone che hanno altri deficit, come disturbi visivi (ad esempio, cataratta), perdita sensoriale (ad esempio neuropatia), danni cerebrali (ad es cerebellare o danno cerebrale a causa del tumore o intervento chirurgico), o scarso adattamento (ad esempio a causa dell'età avanzata) non possono mai ottenere completo ritorno di equilibrio.
In una recente riunione presso la ANA (neuroma acustico associazione), l'autore di questa pagina ha partecipato ad una sessione di 2 ore al giorno e ha intervistato otto pazienti che hanno avuto vertigini persistente. Come regola generale, i pazienti che avevano vertigini due anni dopo l'intervento, avevano avuto una miscela di una lesione cerebellare e perdita totale vestibolare, associato ad un difficile intervento (di solito per un tumore + 4 centimetri). La risonanza magnetica di due pazienti sono qui sopra riportati. Come regola generale, i pazienti che hanno perso sia la funzione dell'orecchio interno (dal acustico), e la sostanziale funzione cerebellare (da un intervento chirurgico dell’ acustico), avranno una perdita durevole di equilibrio
Immediatamente dopo l'intervento chirurgico, la maggior parte dei pazienti hanno vertigini. Una discussione dettagliata di capogiri, vertigini e squilibrio, prima e dopo l'intervento chirurgico può essere trovato
Mal di testa post-operatorio
Mal di testa significativi possono verificarsi in seguito ad interventi chirurgici per neurinomi dell'acustico (revisione Driscoll e Beatty, 1997). L'incidenza varia molto tra un chirurgo e l'altro e dipende anche dalla scelta dell’approccio chirurgico, ma effettuando una panoramica della letteratura si è riscontrata una incidenza di circa il 20-35%. Di fronte di un'incidenza di circa l'8% dopo radiochirurgia.
Schessel et al (1996) hanno osservato e documentato i l'aderenza dei muscoli del collo, sulla dura , dopo craniotomia e segnalato una diminuzione drastica del mal di testa nei pazienti che avevano craniotomia con la sostituzione del lembo osseo. Allo stesso modo, Harner anche notato una riduzione del di mal di testa nei pazienti nei quali avevano avuto una craniotomia in cui era stato utilizzato metacrilato di metile al posto della sola craniotomia (Harner et al, 1995). Si pensa che Il meccanismo sia dovuto, alla trazione sulla dura da parte dei movimenti dei muscoli del collo. Molti pazienti affetti da questa sindrome avevano montato un aggravamento con la tosse o lo sforzo.
Schessel et al (1996) affermano che, nei pazienti che avevano avuto un intervento chirurgico per via retrosigmoidea , la frequenza di mal di testa era significativamente più elevata rispetto a quelli che avevano avuto un approccio translabirintico. Molti altri gruppi di ricercatori hanno trovato dei dati simili . Schaller e Bauman (2003) hanno riscontrato forti mal di testa che richiedono farmaci giornalmente, accompagnati da una sensazione di incapacità nel 34% dei pazienti dopo 3 mesi dall'intervento per via retrosigmoidea. Essi hanno scoperto che questi mal di testa sono stati associati con meningite asettica e, inoltre, che sono associati all'utilizzo di colla di fibrina e di perforazione in faccia posteriore del condotto uditivo interno. Hanno suggerito che la prevenzione del mal di testa post-operatoria può essere realizzata mediante la sostituzione del lembo osseo, alla fine di un intervento chirurgico, l'uso di duraplastic invece di diretta chiusura durale, ed evitare l'uso di colla di fibrina o di perforazione estesa della faccia posteriore del condotto uditivo interno.
Hanno suggerito che la prevenzione del mal di testa post-operatorio può essere realizzata mediante la sostituzione del lembo osseo, alla fine di un intervento chirurgico, utilizzando duraplastica invece della chiusura diretta con la dura ed evitando l'uso di colla di fibrina o di una perforazione estesa della faccia posteriore del condotto uditivo interno
Attualmente ci sono poche informazioni circa l'incidenza di mal di testa utilizzando il metodo della fossa, ma i pochi dati disponibili indicano una incidenza piuttosto bassa (Driscoll et al, 1997).
Nella gestione della cefalea post operatoria si utilizzano analgesici, miorilassanti, antidepressivi e anticonvulsivanti, in modo simile alla gestione dell'emicrania. I farmaci per l'emicrania e comunque, e I farmaci specifici profilattici per l'emicrania non sono raccomandati nella maggior parte dei casi. Tuttavia, un recente rapporto ha rilevato che sumatriptan (un farmaco per l’ emicrania), migliora il mal di testa in 9 / 10 pazienti che avevano una cefalea post-chirurgica. Ciò riflette probabilmente il fatto che , l'emicrania è una condizione molto comune per la salute, che peggiora con qualsiasi tipo di mal di testa.
Un dolore persistente , da incisione, si può verificare per L’intrappolamento del nervo occipitale o dalla formazione di un neuroma occipitale. I massaggi, il calore locale, e gli analgesici possono aiutare. Blocchi del nervo occipitale (come avviene nella clinica del dolore) possono anche essere utile.
Un altro meccanismo che è stato suggerito è che, la polvere ossea, intrappolata all'interno della cavità intracranica possa causare una risposta infiammatoria protratta con conseguente mal di testa cronici (Driscoll, 1997). Le immagini di RM talvolta mostrano un accrescimento della dura (la membrana che ricopre il cervello "sale sopra") e le immagini TC possono mostrare calcificazioni lungo il tronco cerebrale (Schaller e Bauman, 2003). Questo tipo di mal di testa non dovrebbe rispondere al blocco dei nervi del cuoio capelluto (come avviene nella clinica del dolore), e tale procedura può essere di qualche utilità diagnostica. In questi pazienti, logicamente, il trattamento potrebbe includere gli agenti anti-infiammatori ed eventualmente corticosteroidi. Analgesici narcotici sono talvolta indicati..
RM Post-operatoria
Non esistono norme in materia di follow-up del paziente dopo resezione completa del neuroma acustico. In media, però, 3-6 scansioni sono comuni è comune nel corso di un follow-up di circa 5 anni. (Lee e Isaacson, 2005).
Impianti Cocleari
Molto raramente, una persona con un neuroma acustico potrebbe desiderare un impianto cocleare. Ciò potrebbe verificarsi se un tumore acustico è presente nel solo orecchio funzionante o dopo una chirurgia che abbia rimosso un neurinoma acustico bilateralmente. Belal (2001) ha riferito che l'impianto cocleare è possibile solo se vi è un nervo cocleare intatto (come mostrato da una risposta positiva alla stimolazione promontorio), e se l'impianto è fatto al momento della rimozione del tumore acustico, prima della ossificazione della coclea. .
Links:
- Acoustic Neuroma Association
- Support Groups (Acoustic Neuroma Association)
- Presentation by Dr. Hain at ANA association meeting 2009 on balance issues pre and post surgery
References: Timothy C. Hain, MD
·Acoustic Neuroma. NIH Consensus Statement Online 1991 Dec 11-13 ;9(4):1-24
· Anderson, T. D., L. A. Loevner, et al. (2000). "Prevalence of unsuspected acoustic neuroma found by magnetic resonance imaging." Otolaryngol Head Neck Surg 122(5): 643-646.
· Asthagiri AR, Vasquez RA, Butman JA, Wu T, Morgan K, Brewer CC, King K, Zalewski C, Kim HJ, Lonser RR.PLoS One. Mechanisms of hearing loss in neurofibromatosis type 2. 2012;7(9):e46132. doi: 10.1371/journal.pone.0046132. Epub 2012 Sep 26.
· Baker R, Stevens-King A, Bhat N and Leong P (2003). "Should patients with asymmetrical noise-induced hearing loss be screened for vestibular schwannomas?" Clin Otolaryngol 28(4): 346-51.
· Barker FG, 2nd, Carter BS, Ojemann RG, Jyung RW, Poe DS and McKenna MJ (2003). "Surgical excision of acoustic neuroma: patient outcome and provider caseload." Laryngoscope 113(8): 1332-43.
· Belal A. Is cochlear implantation possible after acoustic tumor removal ? Otol Neurotol 22:497-500, 2001.
· Brors D, Schafers M, Bodmer D, Draf W, Kahle G, Schick B. Postoperative magnetic resonance imaging findings after transtemporal and translabyrinthine vestibular schwannoma resection. Laryngoscope 2003 Mar;113(3):420-6
· Chee GH, Nedzelski JM and Rowed D (2003). "Acoustic Neuroma Surgery: The Results of Long-term Hearing Preservation." Otol Neurotol 24(4): 672-6.
· Driscoll CLW, Beatty CW. Pain after acoustic neuroma surgery. Otolaryngologic clinics of North America, vol 30, #5, 1997, 893-903
· Daniels and others. Causes of unilateral sensorineural hearing loss screened by high-resolution fast spin echo magnetic resonance imaging: review of 1070 consecutive cases. Am. J. otol 21:173-180, 2000
· Evans, D. G., A. Moran, et al. (2005). "Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought." Otol Neurotol26(1): 93-7.
· Falcioni M, Taibah A, Di Trapani G, Khrais T, Sanna M. Inner ear extension of vestibular schwannomas. Laryngoscope. 2003 Sep;113(9):1605-8.
· Haggard M, Gatehouse S, Davis A. The high prevalence of hearing disorders and its implications for services in the UK. Brit J. Audiol 1981;15:241-51
· Harner SG, Beatty CW, Ebersold MJ. Headache after acoustic neuroma excision. Am J. Otolaryngol 14:552, 1993
· Hegarty JL. Distal anterior inferior cerebellar artery syndrome after acoustic neuroma surgery. Otol Neurotol 23:560-571, 2002
· Hoistad DL and others. Update on conservative management of acoustic neuroma. Otol Neurotol 22:682-685, 2001
· House JW, Bassim MK, Schwartz M. false-positive magnetic resonance imaging in the diagnosis of vestibular schwannoma. Otology and Neurotology 29:1176-1178, 2008
· Hulshof JH, Hilders CGJM, Barrsma EA. Vestibular investigations in acoustic neuroma. Acta Otolaryngol (stockh) 198, 108; 38-44
· Tanbouzi Husseini S, Piccirillo E, Taibah A, Paties CT, Rizzoli R, Sanna M Malignancy in vestibular schwannoma after stereotactic radiotherapy: A case report and review of the literature. .Laryngoscope. 2011 May;121(5):923-8. doi: 10.1002/lary.21448.
· Jackler RF. Acoustic Neuroma (chapter 21) in Neurotology (Eds Jackler and Brackman, Mosby, 1994).
· Jackler RF. Diagnosing Acoustic Neuromas. Audio-Digest Otolaryngology 33, 04, Feb 21, 2000
· Jackler RF. Acoustic neuromas: selective management Audio Digest Otolaryngology, April 7, 2007.
· Kaplan DM, Hehar SS, Tator C, Guha A, Laperriere N, Bance M, Rutka JA. Hearing loss in acoustic neuromas following stereotactic radiotherapy..J Otolaryngol. 2003 Feb;32(1):23-32.
· Kaylie and others. Acoustic neuroma surgery outcomes. Otol Neurotol 22:686-689, 2001
· Kennedy RJ and others. Intralabyrinthine schwannomas" diagnosis, management, and a new classification system. Otol Neurotol 25:166-167, 2004.
· Komatsuzaki A, Tsunoda A. Nerve origin of the acoustic neuroma. J Laryngol Otol 2001 May;115(5):376-9
· Khrais T, Romano G, Sanna MJ . Nerve origin of vestibular schwannoma: a prospective study..Laryngol Otol. 2007 Nov 27;
· Lee, W. J. and J. E. Isaacson (2005). "Postoperative imaging and follow-up of vestibular schwannomas." Otol Neurotol26(1): 102-4.
· Levo H, Blomstedt G, Hirvonen T, Pyykko I I. Causes of persistent postoperative headache after surgery for vestibular schwannoma. Clin Otolaryngol 2001; 26: 401-406.
· Levo H, Blomstedt G, Pyykko I. Postural stability after vestibular schwannoma surgery. Ann Otol Rhinol Laryngol 2004;113:994-9.
· LIMB CJ, Long DM, Niparko JK. Acoustic Neuromas after Failed Radiation Therapy: Challenges of Surgical Salvage. Laryngoscope 2005;115:93-98.
· Lin VY, Stewart C, Grebenyuk J, Tsao M, Rowed D, Chen J, Nedzelski J. Laryngoscope. Unilateral Acoustic Neuromas: Long-Term Hearing Results in Patients Managed with Fractionated Stereotactic Radiotherapy, Hearing Preservation Surgery, and Expectantly.2005 Feb;115(2):292-296.
· Lin, D., J. L. Hegarty, et al. (2005). "The prevalence of "incidental" acoustic neuroma." Arch Otolaryngol Head Neck Surg 131(3): 241-244.
· Markou K, Eimer S, Perret C, Huchet A, Goudakos J, Liguoro D, Franco-Vidal V, Maire JP, Darrouzet VUnique case of malignant transformation of a vestibular schwannoma after fractionated radiotherapy. .Am J Otolaryngol. 2011 Jun 20.
· Mass S, Wiet RJ, Dinces E. Complications of the translabyrinthine approach for removal of acoustic neuromas. Arch Otolaryngol HNS 1999:125:801-804
· Massuda A and others. Hearing changes after diagnosis in neurofibromatosis type 2. Otol Neurotol 25:150-154, 2004
· Mayo clinical update. Volume 14, #2, 1998.
· Morrison GA, Sterkers JM. Unusual presentations of acoustic tumors. Clin Otolaryngol 1996, 21(1) 80-3.
· Muscat JE and others. Handheld cellular phones and risk of acoustic neuroma. Neurology 2002:58:1304-06
· Nageris BI, Popovtzer A. Acoustic neuroma in patients with completely resolved sudden hearing loss. Ann Otol Rhinol Laryngol 2003 May;112(5):395-7
· Neff B, Willcox T, Sataloff R. Intralabyrinthine schwannomas. Otol Neurotol 24: 299-307, 2003
· Nikolopoulos TP, O'Donoghue Gm. Acoustic neuroma management: an evidence-based medicine approach. Otol Neurotol 23:534-41, 2002
· Noren G and others. Gamma knife surgery in acoustic neuroma. Acta Neurochirg Suppl 1993:58:104-7
· Perry BP, Gantz BJ, Rubinstein JT. Acoustic neuromas in the elderly. Otol Neurotol 22: 389-391, 2001
· Piazza F and others. Management of acoustic neuromas in the elderly: Retrospective study. ENT journal, May 2003, 374-378
· Pollock BE, Lunsford LD, et al. Outcome analysis of acoustic neuroma management: a comparison of microsurgery and stereotactic radiosurgery. Neurosurgery 1995:36:215-229.
· Pothula VB, Lesser T, Mallucci C, May P, Foy P. Vestibular schwannomas in children. Otol Neurotol 22:903-907, 2001
· Ruckenstein MJ, and others. Pain subsequent to resection of acoustic neuromas via suboccipital and translabyrinthine approaches. Am J. Otology 17:620-624, 1996
· Sanna M and others. Treatment of residual vestibular schwannoma. Otol Neurotol 23:980-987, 2002
· Schaller B, Baumann A Headache after removal of vestibular schwannoma via the retrosigmoid approach: A long-term follow-up-study. Otolaryngol Head Neck Surg 2003 Mar;128(3):387-95
· Schessel DA, Nedzelski JM, Rowed D, et al. Pain subsequent to resection of acoustic neuromas via suboccipital and translabyrinthine approaches. Am J Otol 17:620, 1996
· Shin YJ et al. Effectiveness of conservative management of acoustic neuromas. AM J. Otol 21:857-862, 2000
· Slattery WH, Francis S, House KC. Perioperative mortality of acoustic neuroma surgery. Otol Neurotol 22:895-902, 2001
· Soumekh B, Levine S, Haines SJ, Wulf J. Retrospective study of postcraniotomy headaches in suboccipital approach: diagnosis and management. Am J. Otology 1996:17:617-619
· Stewart, T. J., J. Liland, et al. (1975). "Occult schwannomas of the vestibular nerve." Arch Otolaryngol 101(2): 91-95.
· Stidham KR, Robserson JB. Hearing improvement after middle fossa resection of vestibular schwannoma. Otol Neurotol 22:917-921, 2001
· Tos M, Thomsen J, Harmsen A. Is preservation of hearing in acoustic neuroma worthwhile ? Acta Otolarygol (Stockh) 1988: 452: 57-68
· Tufarelli D, Meli A, Labini FS, Badaracco C, De Angelis E, Alesii A, Falcioni M, Sanna M. Balance impairment after acoustic neuroma surgery. Otol Neurotol. 2007 Sep;28(6):814-21.
· UNGER F, Dominikus K, Haselsberger K. [Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas.] HNO. 2010.
· Wackym PA and others. Gamma knife radiosurgery for acoustic neuromas performed by a neurotologist: early experiences and outcomes. Otol Neurotol 25:752-761, 2004
· Warrick P, Bance M, Rutka J. The risk of hearing loss in nongrowing, conservatively managed acoustic neuromas. Am J Otol 20:758-762,
· Wiet RJ, Mamikoglu B, Odom L, Hoistad DL. Long-term results of the first 500 cases of acoustic neuroma surgery. Otolaryngol Head Neck Surg 2001; 124: 645-51. (Comment: Note that Dr. Wiet sees patients in the same location as Dr. Hain, the author of this review)
|
BIBLIOGRAFIA NEURINOMI: Gruppo Otologico Sanna 1) Diagnosi radiologica del neurinoma dell'acustico intracanalare: la cisternografia opaca è ancora giustificata? (in coll. con P. Bassi, C. Zini, G. Jemmi) Atti 69° Congr. Soc. Ital. O.R.L., Roma 19-23 Maggio 1982. 2) Hearing preservation in acoustic neuroma surgery: middle fossa versus sub occipital approach. (in coll. con C. Zini, A. Mazzoni, A. Gandolfi, R. Pareschi, E. Pasanisi, R. Gamoletti) Am. J. Otol. 8, 500-506, 1987. 3) Hearing preservation in acoustic neuroma surgery: middle fossa vs. sub occipital approach. (in coll. con C. Zini, A. Mazzoni, A. Gandolfi, R. Pareschi, R. Pasanisi, R. Gamoletti) Clinical Audiology 87. 4) Management of facial nerve in acoustic tumors surgery. (in coll. con A. Gandolfi, A. Mazzoni, C. Zini, E. Pasanisi) Atti International Conference on: 7) Hearing preservation: a criticai review of the literature. (in coll. con C. Zini, R. Gamoletti, M. Landolfi, M. Shaan, F. Piazza) Acoustic Neuroma, 1992 8) Growth rate of acousdc neuromas. (in coll. con A.K. Taibah, M.Landolfi, L. Vassalli, A. Russo, E. Pasanisi, M. Shaan)Acoustic Neuroma, 1992 Proceedings of the First International Conference Copenhagen, Denmark, August 25-29, 1991 Kugler and Ghedini Pub., Amsterdam, 183-186, 1992. 9) Preservation of the facial nerve in acoustic neuroma Surgery via the translabyrinthine approach. (in coll. con A.K. Taibah, M. Landolfi, L. Vassalli, A. Russo, E. Pasanisi, M. Shaan) Acoustic Neuroma, 1992 Proceedings of the First International Conference Copenhagen, Denmark, August 25-29, 1991 Kugger and Ghedini Pub., Amsterdam, 389-390, 1992.
11) Translabyrinthine removal of a very large acoustic neuroma. (in coll. con A. Russo, A.K. Taibah, M. Landolfi, L. Vassalli, E. Pasanisi, G. Chelmis) Acoustic Neuroma,1992 Proceedings of the First Internadonal Conference Copenhagen, Denmark, August 25-29,1991 Kugler and Ghedini, Amsterdam, 393-395, 1992. 13) The pors and cons of the translabyrinthine, middle cranial fossa and retrosigmoid approaches in the of acoustic neuromas. (in coll. con C. Zini, A. Gandolfi, F. Piazza) Acoustic Neuroma, 1992 Proceedings of the First International Conference Copenhagen, Denmark, August 25-29, 1991 Kugler and Ghedini Pub., Amsterdam, 499-500, 1992 14) The problem of facial nerve anatomical and functional preservation in relation to different approaches(in coll. con F. Piazza, C. Zini, C. Iagher) Acoustic Neuroma, 1992 Proceedings of the First International Conference Copenhagen, Denmark, August 25-29, 1991 Kugler and Ghedini Pub., Amsterdam, 767-770, 1992 15) Rerouting and end-to-end anastomosis of the facial nerve in translabyrinthine removal of acoustic neuromas. (in coll. con L. Vassalli, A.K. Taibah, M. Landolfi, A. Russo, E. Pasanisi, G. Chelmis) Acoustic Neuroma, 1992 Proceedings of the First International Conference Copenhagen, Denmark, August 25-29, 1991 Kugler and Ghedini Pub., Amsterdam, 775-776, 1992 16) Removal of a meningioma of the cerebellopontine angle via the extended translabyrinthine approach. (in coll. con M. Landolfi, L. Vassalli, A.K. Taibah, A. Russo, E. Pasanisi, G. Chelmis) Acoustic Neuroma, 1992 Proceedings of the First International Conference Copenhagen, Denmark, August 25-29,1991 Kugler and Ghedini Pub., Amsterdam, 965-968,1992 17) Synopsis on: Hearing preservation following acoustic neuroma surgery. 18) Atypical preservation of acoustic neuroma(in coll. con M. Shaan, L. Vassalli, M. Landolfi, A. Taibah, A. Russo) Otolaryngol. Head Neck Surg., 109, 865-870, 1993.159) Decision making in acoustic neuroma management: the only hearing ear. (in coll. con M. Naguib, E. Saleh, M. Aristegui, A. Mazzoni) Skull Base Surgery 4, 1, 43-47, 1994. 19) Management of the high jugular bulb in the translabyrinthine approach. 20) The enlarged translabyrinthine approach for removal of large vestibular schwannomas. (in coll. con M. Naguib, E. Saleh, Y. Cokkeser, M. Aristegui, M. Landolfi, A. Taibah, A. Mazzoni) Journ. Laryng. Otol. 108, 545-550,1994. 21) Via della fossa cranica media allargata: una analisi morfometrica. (in coll. con M. Landolfi, M. Aristegui, A. Taibah, A. Russo, E. Saleh) Acta 22) Management of the high jugular bulb in the translabyrinthine approach. (in coll. con E. Saleh, M. Aristegui, A. Taibah, A. Mazzoni) Otolaryngol. Head 195) Incidenza della normacusia nel neurinoma dell'acustico. (in coll. con G. De Donato, A. Russo, E. Saleh) Acta Otorhinol. Ital. 15, 1995. 23) Measurement of endocochlear DC potentials in ears with acoustic neuromas: a preliminary report. (In coll. con T. Kobayashi, A. Aslan, T. Chiba, T. Takasaka) Acta Otolaryngol. (Stockh) 116, 791-795,1996. 24) Vestibular schwannoma and the only hearing ear. (In coll. con S. Bhatia, S. Karmarkar, A. Taibah, A. Russo) The Journal of Laryng. and Otol. 110, 366- Proceedings of the 2nd International Conference on Acousdc Neuroma Surgery and 2nd European Skull Base Society Congress, Paris, France, April 22-26, 1995. Edited by J. M. Sterkers, R. Charachon and 0. Sterkers 26) Management of acoustic neuroma through the enlarged translabyrinthine approach. How can we improve the results ? (In coll. con F. Mancini, M. Falcioni, M. Landolfi, A. Taibah, A. Russo, A. Mazzoni) Acoustic Neuroma and Skull Base Surgery, pp. 185-193Proceedings of the 2nd International Conference on Acoustic Neuroma Surgery and 2nd European Skull Base Society Congress, Paris, France, April 22-26,1995. Edited by J. M. Sterkers, R. Charachon and O. Sterkers 27) Normal hearing in acoustic neuroma. (In coll. con M. Falcioni, G. De Donato, E. Saleh, A. Taibah, A. Russo) Acoustic Neuroma and Skull Base Surgery, pp. 83-86.Proceedings of the 2nd International Conference on Acoustic Neuroma Surgery and 2nd European Skull Base Society Congress, Paris, France, April 22-26, 1995. 28) Normal hearing in acoustic neuroma patients: a critical evaluation. (In coll. con E. Saleh, M. Aristegui, M. Naguib, Y. Cokesser, M. Landolfi) The 29) La liquorrea come complicanza della via translabirintica nel trattamento del neurinoma dell'acustico. (In coll. con Falcioni M., Taibah A., De Donato G., Sanna M.) Acta Otorhinolaryngol. Ital. 18, 63-69,1998 30) Coesistenza di un neurinoma dell'acustico e di un tumore glomico timpanico. 31) Identificazione del nervo facciale nella via translabirintica: un metodo alternativo. (in coll. con Falcioni M, De Donato G, Piccirillo E, Taibah A, Russo A, Mancini F, Caruso A, Piccirillo E.) Acta Otorhinol. Ital. 19; 1-5, 1999 32) Transpetrous removal of residual vestibular schwannoma. 33) Translabyrinthine vs retrosigmoid approach for small tumors.(in coll. con Bacciu A, Bruzzo M, Falcioni M, Chays A, Magnan J.) 34) Technical refinements in translabyrinthine approach for acoustic tumor removal. (in coll con Aristegui M, Falcioni M, De Donato G, Taibah A, Russo A, Mancini F.) Third International Conference on Acoustic Neurinoma and Other C.P.A. Tumors. Rome, 12 to 17 June 1999, CIC international editions. 35) Facial nerve in translabyrinthine approach: an alternative technique. 37) Complications in translabyrinthine approach. (in coll. con De Donato G, Taibah A, Russo A, Mancini F, Falcioni M, Piccirillo E.) 38) Hospital stay in CPA surgery. (in coll. con Piccioni L, Taibah A, Mancini F, Falcioni M, De Donato G, Piccirillo E.) Third Intemational Conference on Acoustic Neurinoma and Other C.P.A. Tumors. Rome, 12 to 17 June 1999, CIC intemational editions. 39) Decision making in acoustic neuroma of only hearing ear. 40) Normal hearing and sudden hearing loss in acoustic neurinoma. 42) Il trattamento dei neurinomi dell'acustico residui. (in coll. con Falcioni M, Piccioni L, Taibah A, De Donato G, Russo A, Piccirillo E.) Acta Otorhinol. Ital. 20; 151-8; 2000. 43) Hearing preservation in vestibular schwannoma surgery: fact or fantasy. 45) Arachnoid cysts of the petrous apex in a patient with vestibular schwannoma. 46) Electromyographic evaluation of facial nerve damage in acoustic neuroma surgery. (in coll. con Nakao Y, Piccirillo E, Falcioni M, Taibah A, Kobayashi T.) 47) La via translabirintica allargata nei neurinomi dell'acustico di grandi dimensioni. (in coll. con Falcioni M, Russo A, Mancini F, Taibah A, Piccioni L, De Donato G, Caruso A.) Acta Otorhinol. Ital. 21, 226-236, 2001. 48) Identification of the facial nerve in the translabyrinthine approach: an alternative technique. (in coll con Saleh E, Russo A, Falcioni M.) Otolaryngol. Head Neck Surg. 124, 105-6, 2001. 49) Electromyographic evaluation of facial nerve damage in acoustic neuroma surgery. (in coll. con Nakao Y, Piccirillo E, Falcioni M, Taibah A, Kobayashi T.) 50) Treatment of residual vestibular schwannoma. (in coll. con Falcioni M., Taibah A.K., De Donato G., Russo A. Piccirillo E.) Otology and Neurotology 2002. 51) Prediction of facial nerve outcome using electromyographic responses in acoustic neuroma surgery. (in coll. con Nakao Y, Piccirillo E, Falcioni M, Taibah A, Russo A, Kobayashi T.) Otol. & Neurotol. 23, 93-95, 2002. 52) Residual vestibular schwannomas treatment. (in coll. con Falcioni M, Taibah A, De Donato G, Russo A, Piccirillo E.) Otol. & Neurotol. 23: 980-987; 2002 53) Neurinoma dell'acustico ipervascolarizzato. (in coll. con Di Trapani G, Falcioni M, Taibah A, Russo A.) ORL up-to-date. 89° Congresso Nazionale della 55) Hearing preservation surgery in vestibular schwannomas. (in coll. con Piccirillo E, Russo A, Khrais T, Rohit, Falcioni M.) IVth international conference 57) CSF Teak after vestibular schwannoma surgery. (in coll. con Mancini F, Khrais T, Falcioni M, Di Trapani G.) IVth international conference on vestibular shwannoma and other CPA lesions. Cambridge UK, 13th-17 th July 2003. Baguley D, Ramsden R and Moffat D eds. Immediate proceedings LDT, 2001. 58) Enlarged translabyrinthine approach for the management of large and giant acustic neuromas (vestibular schwannomas). A report of 175 consecutive cases. 59) Perioperative complications in acoustic neuroma ( vestibular schwannoma) surgery. (in coll. con Taibah A., Russo A., Falcioni M., Agarwal M.) Otology and 60) Facial nerve grafting in the cerebellopontine angle. ( in coll con Yogesh J., Falcioni M., Mancini F., Romano G.) The Laryngoscope 114: April 2004 62) Hearing preservation surgery in vestibular schwannoma: the hidden truth. (in coll con: Khrais T., Russo A., Piccirillo E., Augurio A.) Annals of Otology, 64) [Clinical experience in 36 cases of using of the extended translabyrinthine technique for the treatment of large acoustic neuromas] 65) Preoperative predictive factors for hearing preservation in vestibular schwannoma surgery. Ann Otol Rhinol Laryngol. 2006 Jan;115(1):41-6. (in coll con Rohit, Piccirillo E, Jain Y, Augurio A). 66) Hearing preservation surgery in vestibular schwannoma. 67) Quality of life after acoustic neuroma surgery. Otol Neurotol. 2006 Apr;27(3):403-9. (in coll con Tufarelli D, Meli A, Alesii A, De Angelis E, Badaracco C, Falcioni M) 68) Intracanalicular meningioma: clinical features, radiologic findings, and surgical management. Otol Neurotol. 2007 Apr;28(3):391-9. (in coll con Bacciu A, Piazza P, Di Lella F). 69) Management of intralabyrinthine schwannomas. Auris Nasus Larynx. 2007 Dec;34(4):459-63. Epub 2007 Apr 27. (in coll con Di Lella F, Dispenza F, De Stefano A, Falcioni M) 70) Intraoperative cochlear nerve monitoring in vestibular schwannoma surgery--does it really affect hearing outcome? Audiol Neurootol. 2008;13(1):58-64. Epub 2007 Sep 20. in coll con Piccirillo E, Hiraumi H, Hamada M, Russo A, De Stefano323) Cerebrospinal Fluid Leak After Retrosigmoid Excision of Vestibular Schwannomas. Otol Neurotol. 2007 Dec 28; (in coll con Maurizio Falcioni, Romano G, Nitin Aggarwal) 71) Nerve origin of vestibular schwannoma: a prospective study. J Laryngol Otol. 2007 Nov 27;:1-4(in coll con Khrais T, Romano G) 327) Decision making for solitary vestibular schwannoma and contralateral Meniere's disease. Dispenza F, De Stefano A, Flanagan S, Romano G, Sanna M. 72) Intraoperative cochlear nerve monitoring in vestibular schwannoma urgery--does it really affect hearing outcome? Piccirillo E, Hiraumi H, Hamada M, Russo A, De Stefano A, Sanna M. Audiol Neurootol. 2008;13(1):58-64. Epub 2007 Sep 20.
|
















 L'esposizione prolungata a dei livelli sonori eccessivi può essere fonte di lesioni uditive irreversibili con un aumento permanente delle soglie uditive (permanent threshold shift [PTS]). Dal momento che i livelli sonori e la durata di esposizione sono interdipendenti, si ammette che la gravità delle perdite uditive definitive è legata alla quantità di energia acustica ricevuta dall'orecchio. La normativa ISO 1999: 1990 [
L'esposizione prolungata a dei livelli sonori eccessivi può essere fonte di lesioni uditive irreversibili con un aumento permanente delle soglie uditive (permanent threshold shift [PTS]). Dal momento che i livelli sonori e la durata di esposizione sono interdipendenti, si ammette che la gravità delle perdite uditive definitive è legata alla quantità di energia acustica ricevuta dall'orecchio. La normativa ISO 1999: 1990 [


![[clip_image002%255B4%255D.jpg]](/images/IPOACUSIA_PROGRESSIVA_SU_BASE_GENETICA_file/image003.jpg)