Vertigini dell’infanzia
- Categoria: QUADRI CLINICI OTONEUROLOGICI DA ALTERAZIONE/INSUFFICIENZA DEL CIRCOLO VERTEBRO-BASILARE
- Pubblicato: Giovedì, 17 Novembre 2016 09:26
- Visite: 16002
Vertigini dell’infanzia- del bambino
Parole chiave : Vertigini, Vertigine benigna parossistica, Disturbo di vergenza, Problemi oculari, Equilibrio, Neurite, Labirintite, Frattura della rocca
· Introduzione
· Il sintomo: vertigine
Che cos’è che la vertigine?
Donde vengono le sensazioni di movimento e la sensazione
vertiginosa?
· Esame clinico oto-neuro-vestibolare del bambino con vertigini 2
Esame vestibolare clinico di base
Esame neurologico clinico
Esame oftalmologico
· Esami vestibolari
Test della funzione canalare
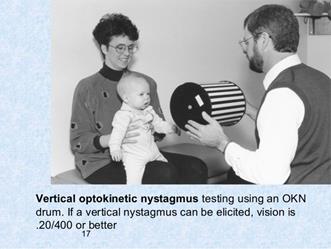
Test della funzione otolitica
· Quali sono le cause di vertigine nel bambino?
Equivalenti emicranici
Vertigine parossistica benigna del bambino (20%)
Quadro di trauma cranico (10%)
Malformazione dell’orecchio interno (malformazione di Mondini)
Disturbi visivi
Neurite vestibolare
Labirintite
Tumore della fossa posteriore
Altre diagnosi
● Conclusione
Riassunto
Le vertigini e i disturbi dell'equilibrio del bambino, quando sono riconosciuti, allarmano i medici e le famiglie e conducono con troppa frequenza a indagini inutili e costose ancor prima di un buon esame clinico oto-neuro-vestibolare. Per la redazione di questo capitolo si sono utilizzati i risultati di uno studio condotto per 14 anni su più di 2.000 pazienti inviati per vertigini o disturbi dell'equilibrio all'unità di esplorazioni funzionali vestibolari del reparto di otorinolaringoiatria pediatrica dell'ospedale Robert Debré (Parigi). In questa sede verranno trattati i principali segni che evocano un disturbo dell'apparato dell'equilibrio nonché le principali eziologie riscontrate nel bambino (equivalenti emicranici, disturbi oftalmologici, vertigine parossistica benigna del bambino e trauma). Ciò ha permesso di definire la migliore condotta diagnostica e terapeutica da tenere di fronte a una vertigine del bambino.
La Vertigine nei bambini è una diagnosi difficile da fare e da valutare. Le cause di vertigine nei bambini differiscono da quelli osservati negli adulti e la sua presenza può essere facilmente trascurata o attribuita a loro statue di sviluppo e mancanza di coordinamento. I bambini potranno comunemente presenti con la denuncia principale di vertigine che può essere suddiviso in quattro categorie:. Vertigine, presincope, squilibrio, e sensazione di testa vuota vera vertigine è definita come l'illusione del movimento, di solito la rotazione, anche se spostamento lineare o inclinazione può anche essere sperimentato. vera vertigine può essere ulteriormente suddivisa in vertigine soggettiva in cui il paziente sperimenta l'illusione del movimento in relazione con l'ambiente e la vertigine oggettiva in cui l'ambiente è sentita muoversi rispetto ad un paziente stazionaria. Vertigo è in genere accompagnata da sintomi quali nausea, vomito, pallore, sudorazione e senza perdita di coscienza. La patologia responsabile di vertigine può risiedere in diverse località, con il sistema vestibolare più importante tra di loro. Danni ai sistemi visivi e propriocettivi può anche essere responsabile oltre a più lesioni centrali del sistema nervoso centrale. Questi siti di patologia possono essere suddivisi nelle vertigine periferica o quello derivante dal sistema vestibolare nell'orecchio interno e vertigini centrali connessi al resto del sistema nervoso. Anche se i disturbi vertiginosi si è trovano più comunemente nella popolazione adulta e anziana, si verifica ancora abbastanza frequentemente nei bambini per giustificarne una valutazione . L'esperienza di vertigine può essere difficile da descrivere per un adulto, perché molti dei segnali fisiologici periferici del sistema vestibolare, il sistema visivo, gli ingresso somatosensoriali ed i sistemi propriocettivi si verificano al di sotto delle soglie di sensazione Hullar et al.,2005. La storia di bambini più giovani è ulteriormente ostacolata dalla immaturità del loro capacità di comunicazione e la necessità di fare affidamento contare su una squadra secondaria, i loro genitori , per descrivere i loro episodi Inoltre, disfunzioni vestibolari possono presentarsi in modo diverso in bambini rispetto agli adulti: si possono presentare come disturbi della vista, mal di testa, andatura instabile o goffa, ritardo motorio, difficoltà di apprendimento e / o attacchi di paura inspiegabili. Inoltre, i bamb ini con disfunzioni vestibolari congeniti o compensato possono essere senza sintomi. O ’Reilly , et al., 2010; Worden et al.,2007 Weiss AH, Philips JO.,2006;La sensazione vertiginosa, ovvero l'informazione erronea di movimento, può essere dovuta, nel bambino come nell'adulto, a un'anomalia del funzionamento dei tre grandi sistemi sensoriali che forniscono questa informazione, vale a dire il sistema visivo, il sistema vestibolare e il sistema propriocettivo-somestesico. La vertigine nel bambino, quando è riconosciuta, è un sintomo che getta nel panico in generale le famiglie e i medici. Le vertigini e i disturbi dell'equilibrio conducono allora spesso a un eccesso di prescrizione di esplorazioni funzionali inutili e costose (risonanza magnetica [RM], TC) senza contribuire a una gestione terapeutica adeguata. Ciò è dovuto in gran parte all'ansietà proveniente dall'equivocare una condotta diagnostica chiara, che ha lo scopo di non mancare la diagnosi di «tumore della fossa posteriore», diagnosi molto temuta e, in realtà, rara (meno dell'1% delle vertigini del bambino). Davanti alle d vertigini el bambino, cosa si deve fare? Questa è una domanda importante.
Le sensazioni vertiginose sono un sintomo che non è facile da riconoscere nel bambino e molti medici ritengono che siano rare. L'incidenza delle vertigini è in effetti sottostimata per diverse ragioni:
• uno scarso riconoscimento del sintomo: la vertigini può passare inosservata nei bambini molto piccoli che non possono descrivere la loro sensazione vertiginosa e in cui sono solamente evidenti i segni associati alle vertigini i (atassia, vomito, pallore, dolori addominali), il che orienta il medico verso patologie intestinali (gastroenteriti per esempio) o neurologiche (tumori della fossa posteriore) e non lo porta a effettuare gli esami adeguati;
• un mancato riconoscimento delle diverse cause di vertigine e della loro frequenza è legato a una scarsa conoscenza del sistema vestibolare e della sua importanza nel controllo posturale e motorio del bambino. Questo è anche conseguenza del fatto che per lungo tempo non sono stati disponibili test, adatti ai bambini piccoli, per esplorare la funzione vestibolare nel suo insieme;
• l'assenza di un'attitudine diagnostica coerente che tenga conto della frequenza di ogni patologia pediatrica che si esprime attraverso le vertigini in funzione dell'età, e la scarsa conoscenza di esami clinici semplici che permettono di orientare la diagnosi e dei rischi e costi dei differenti esami richiesti. Ciò ha lasciato e lascia ancora una gran numero di vertigini del bambino senza alcuna diagnosi eziologica precisa e senza trattamento adatto.
Rassegna della letteratura
Gli effetti della maturazione delle risposte per-rotatoria nei bambini sono stati particolarmente studiati nel 1970 (Eviatar, Eviatar, e Naray, 1974; Eviatar, Miranda, Eviatar, Freeman, e Borkowski, 1978). Sebbene i neonati a termine dimostrano una risposta nystagmic forte di coorti premature, le risposte sembrano essere comparabili di nove mesi di età. Effetti dell'età su riflesso vestibolo-oculare (VOR) a seguito di irrigazione calorica è stata studiata anche da numerosi ricercatori con risultati variabili. Van der Laan e Oosterveld (1974) hanno trovato nei bambini rispetto agli adulti: una frequenza della risposta nistagmica meno intensa ma con ampiezze superiori con,. Krejčová e colleghi (1975), tuttavia, al contrario hanno riferito che i bambini mostrano una frequenza di battiti maggiore rispetto agli adulti. Andrieu-Guitrancourt et al. (1981) hanno scoperto che la frequenza dei battiti aumenta man mano che il bambino matura, mentre la velocità massima dell'occhio diminuisce. In alcuni studi si è trovato che le misure di velocità ed ampiezza degli occhi sono più intense nei bambini rispetto a adolescenti (Ornitz, et al., 1979) mentre altri ricercatori hanno osservato la risposta più intensa in età adulta (Pacciame e Peterman, 1979). Il VOR passa attraverso diverse fasi di sviluppo nella prima infanzia (Donat, Donat, e Swe Lay, 1980) e dovrebbe essere considerato anormale se il VOR è assente dall'età di dieci mesi di età (Fife, et al., 2000). Gli esaminatori di riferimento hanno sottolineato l'importanza di considerare queste differenze di età quando si interpretano i risultati dei test pediatrici (Kenyon, 1988; Levens, 1988).
Occasionalmente, i sintomi vestibolari pediatrici sono collegati ad altri disturbi, come l'autismo, ritardo motorio, difficoltà di apprendimento, e dislessia. 1 Nandi e Luxon 20082 riportati i più comuni cause di disfunzione vestibolare nei bambini erano emicrania, otite media, e traumi, e che osservato circa il 30% e il 40% delle persone sorde hanno sistemi vestibolari insoliti. Szirmai 2010 3 ha valutato 145 pazienti vertiginosi fino all'età di 18 anni e ha concluso quasi due terzi di loro hanno dimostrato anomalie vestibolari. Le cause più comuni di vertigini riportato da Szirmai inclusi disturbi "extra-vestibolare" come la malattia di panico e disturbo d'ansia. Tuttavia, per i pazienti più giovani, sintomi vestibolari sono stati più spesso causati da emicrania.
Wiener-Vacher 2008 4 ha riferito circa 2.000 bambini di età superiore ai 14 anni, che sono stati deferiti per problemi di equilibrio e vertigini. Nel suo studio, il 20% dei bambini profondamente sordi avuto la completa perdita vestibolare bilaterale, il 40% ha avuto perdita bilaterale parziale o asimmetrica vestibolare, e il 40% aveva una normale funzione vestibolare bilaterale. Wiener-Vacher bambini determinate di cui a lei per la valutazione dei loro disturbi vertigini o saldo ha la seguente diagnosi (elencati in ordine di prevalenza):
1) Equivalenti emicranici: il 25% dei bambini ha avuto mal di testa che hanno preceduto, accompagnato, o alternati con le vertigini.
2) Vertigine parossistica benigna dell'infanzia: il 20% dei bambini (con l'accento sulla i 2 ei 3 anni) ha riferito breve vertigini (meno di 10 minuti), senza mal di testa.
3) trauma cranico: il 10% dei bambini valutati segnalato vertigini post-trauma cranico. Queste denunce potrebbero essere indicativi di frattura dell'osso temporale o di una fistola perilinfa.
4) Mondini (e altre) malformazioni dell'orecchio interno: Circa il 10% dei bambini della serie avevano malformazioni. È interessante notare che, nella sindrome CHARGE, Wiener-Vacher riporta quasi il 100% dei bambini ha avuto canali semicircolari assenti.
5) Disturbi oftalmici: Circa il 10% di tutti i bambini di cui ha avuto problemi visivi.
6) vestibolare neurite: Si è verificato in circa il 5% dei bambini.
7) tumori della fossa posteriore: si è verificato in meno dell'1% di tutti i bambini di cui.
8) Varie: Rarissimo sono stati casi che coinvolgevano l'origine psichiatrica, ipotensione ortostatica, epilettici, auto-imminue, Vertigine parossistica posizionale benigna (VPPB), la malattia di Meniere, idrope endolinfatico ritardo, ecc Wiener-Vacher 20084
Pertanto, l'analisi delle questioni vestibolari e di equilibrio nei bambini è molto importante per i genitori, il bambino, l'audiologo, e il medico.
Storia / Sintomo Principale
La storia è in genere la chiave per diagnosticare un bambino con un disturbo dell'equilibrio; una storia accurata solo determina spesso l'eziologia di squilibrio del paziente. Non è insolito per l'esame fisico e test per fornire visione poco supplementare nella diagnosi. E 'meglio lasciare prima il paziente e / o il loro tutore per descrivere i loro sintomi in modo aperto con direzione minimo dal medico. La storia può essere notevolmente facilitato da un approfondito previsit questionario compilato dal paziente e / o tutore (vedi Appendice). Il questionario non solo migliora l'efficienza della raccolta dei dati al momento della visita, ma aiuta anche i genitori affinare le loro osservazioni. lt è in genere migliore per avere dei pazienti e / o dei loro tutori compilare il questionario nel comfort della propria casa ben prima della visita clinica. Il questionario può quindi aiutare a guidare l'intervista durante la visita clinica. La storia, una volta completato sia attraverso il questionario e visitare, dovrebbe consentire al medico di avere un senso di se i sintomi siano dovuti ad un'eziologia periferico.
Considerazione Speciale
La storia è in genere la chiave per diagnosticare un bambino con un disturbo dell'equilibrio.
Una volta che il paziente ha descritto i suoi sintomi, la prima distinzione è la differenza tra vertigini e disturbi dell’equilibrio. I disturbi dell’equilibrio SONO spesso ed erroneamente usati per indicare le vertigini. La Vertigine è la sensazione di movimento rotazionale interno anormale o movimenti anomali dell’ ambiente esterno. La radice della parola è basato sul latino, vertere, che significa girare. Non sorprende che, la vertigine è classicamente associata a una sensazione di rotazione illusoria, ma può anche essere presente come movimento anomalo di oscillazione anteroposteriore o anche un cambiamento relativo di orientamento rispetto alla linea verticale Hullar et al.,2005.2 Vertigini è un termine molto più ampio, termine non specifico che descrive l’ interruzione del proprio orientamento conr l'ambiente e include ma non si limita a vertigini, squilibrio, e debolezza.
La pietra angolare della storia è quella di determinare se il bambino sta vivendo vere vertigini e, in caso affermativo, quanto lungo è stato l’ ultimo effettivo episodio vertiginoso, E 'importante ricordare che, dopo un attacco di vertigini, il paziente può avvertire squilibrio intercritico, che si può equiparare come "stato vertiginoso", ma non veramente vertige, i pazienti più anziani si descrivono un senso di distacco o di dissociazione come se fossero in una "nebbia" o come se essi sono in piedi o camminare "sul ponte e sui cuscini di una nave che rolla. "Con i pazienti più giovani, spesso è utile per verificare se le sensazioni che sperimentano sono simili a quelli che hanno dopo aver girato su se stessi o in un parco giochi. Una storia di successo farà la differenza tra un disturbo dell’equilibrio sfumato ed unavera vertigine. Una denuncia otologica unilaterale quando si verifica è un sintomo molto utile localizzante in un paziente vertiginoso: la pienezza sonora, il tinnitus, la perdita dell'udito, o distorsione sonora. Per esplorare questi sintomi, il medico può spesso localizzare il sito della lesione prima dell'esame o testing.5 Sia la presenza e la durata delle vertigini sono fondamentali per poter formulare una diagnosi accurata.
Considerazione Speciale
Una disturbo otologico unilaterale quando si verifica è un sintomo molto utile localizzante in un paziente vertiginoso.
In primo luogo, chiedere al paziente e / o al tutore di descrivere il primo episodio con le loro parole così come un tipico episodio successivo, senza usare la parola vertigini, Sono gli episodi sostenuti o ricorrente? Una volta che un tipico episodio è stato completamente concretizzate nelle parole del paziente, il medico deve chiedere la frequenza, la durata, la prevedibilità,
Fico. 11.1 Pratica percorso: vertigini.
esordio, modificando i fattori, fattori provocanti, e sintomi associati. Nave qualsiasi degli episodi stata associata a nausea, vomito, o, in mancanza?
I pazienti con disturbi labirintici periferici descrivono facilmente le loro sensazioni vertiginose. I pazienti con disfunzione acuta del sistema nervoso centrale (CNS) possono o non possono avere vertigini, considerando che le disfunzione cronica del SNC e cerebrovascolari, cardiovascolari, e le cause metaboliche di vertigini raramente produce sensazioni di moto relativo. Vertigini periferica è disponibile in incantesimi che ultimi secondi di minuti (benigno vertigini parossistica posizionale), minuti ad ore (malattia di Ménière), o ore a giorni (vestibolare neurite). La perdita dell'udito, acufeni e l’ovattamento auricolare sono sintomi frequenti di malattia periferica. E’ importante definire se la perdita di udito era improvvisa o progressiva e se si avevano o meno fluttuazioni dell’udito. I cambiamenti di posizione spesso aggravano lo squilibrio, e la posizione supina riduce i sintomi. Si sospettata Vertigine Parossistica Posizionale Benigna, per esempio, nei pazienti con brevi episodi vertiginosi innescati dai cambiamenti di posizione della testa. nella maggior parte degli episodi, l'esordio è improvviso, anche se l’inizio della sintomatologia è spesso meno definito . I pazienti spesso si sentono bene tra un episodio e l’altro.5
A differenza delle vertigini periferiche, l'espressione sintomatica di cause centrali di vertigini è più variabile. Le sensazioni descritte sono spesso rotazione, inclinazione, la sensazione di essere spinto verso un lato, vertigini, confusione, o anche la sincope. Se è documentata la perdita di coscienza, una eziologia periferica è quasi mai la causa. Utile per la localizzazione centrale è la presenza di segni di accompagnamento di disfunzione neuronale: disartria, disfagia, diplopia, parestesie, emiparesi, grave cefalea localizzata, convulsioni, e la perdita di memoria, la durata dei sintomi è più varia e dura da minuti a ore. Gli effetti dei cambiamento di posizione sono meno prevedibile. Queste costellazioni di sintomi suggeriscono un disturbo del tronco cerebrale o corticale, piuttosto che labirintico, 5
Molti fattori medici ed emotivi possono promuovere un senso di vertigini e squilibrio. Ipertensione, ipotensione, patologie endocrine, e l'ansia può aggravare e/o causare vertigini, sincope e/o instabilità, ma raramente produrre vere vertigini. Questi problemi di salute generale ed emotivo dovrebbero essere esplorati nel corso dell’anamnesi.
Medicazioni e Farmaci
è importante rivedere tutti i farmaci, compresi i farmaci NON PRESI IN CONSIDERAZIONE, e per informazioni sulla possibile uso illecito di droga ed alcool. Ha il paziente mai assunto farmaci ototossici? L'elenco dei farmaci recensiti come possibili ototossici dovrebbe includere, (e non limitarsi alla gentamicina per via endovenosa), la tobramicina, vancomicina, furosemide, l'aspirina, ed il cisplatino. Il questionario previsto è particolarmente utile in questo senso perché i genitori ei pazienti spesso non riescono a ricordare esattamente i farmaci, soprattutto con il passare del tempo.
Passato medico e Storie chirurgiche
È il paziente nato a termine? È stata un parto semplice ? C'è stata l'esposizione a eventuali agenti infettivi: parotite, morbillo, rosolia, citomegalovirus, herpes, l'HIV, la sifilide o la malattia di Lyme? Il paziente ha un’anamnesi(storia) corrente o remoto di malattia dell'orecchio? Vi è mai stata una storia di meningite o accidente cerebrovascolare? Il paziente ha mai sottoposto a chemioterapia e / o radioterapia? Ci sono dei medici di base! condizioni come la malattia della tiroide, diabete mellito, malattie cardiache, malattie renali, malattie oftalmologico, malattia autoimmune, o coagulopatie? Il paziente è stata mai sotto èi anestesia generale e, in caso affermativo, quali operazioni ha subito il paziente?
Anamnesi(Storia) familiare
Vi è una storia familiare di vertigine / disequilibrio? Sia l’ emicrania che la malattia di Ménière possono presentare una tendenza ad essere familiari.
Esame fisico
Con il tempo la storia è completata, il clinico tipicamente ha ridotto la diagnosi differenziale a poche, se non, ad una sola ipotesi diagnostica. Come tale, l'esame fisico può essere diretto con i tests delle ipotesi diagnostiche. Il paziente deve sottoporsi ad un Esame neuro-otologico attento per la certificazione e standardizzato, che comprende un esame approfondito della testa e del collo, sia con la pneumatoscopia o pneumomicrotoscopia e un esame dei nervi cranici. L'esame testa e il collo deve cercare cause comuni che possono esacerbare il senso di disequilibrio come l’occlusione per cerume, l’otite media acuta e l'otite media con effusione, l’otite media suppurativa cronica, così come la sinusite acuta e cronica. Anomalie Congenite del padiglione auricolare, canale uditivo esterno, e della faccia possono essere associati con anomalie del labirinto
Considerazione Speciale
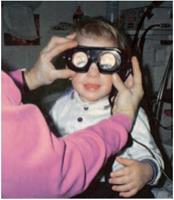
Gli occhiali di Frenzel sono di grande aiuto nella valutazione dei movimenti oculari.
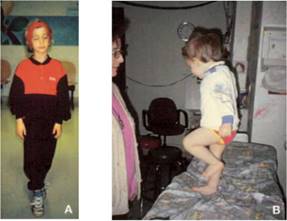
Dopo che è completato l'esame di routine della testa e il collo, il medico valuta attentamente i movimenti oculari ed esegue una breve, esame neurologico diretto. Per la valutazione dei movimenti oculari, le lenti di Frenzel sono di grande aiuto. Le Lenti di Frenzel con lenti di ingrandimento sono illuminati internamente che si confondono del paziente visione impediscono un paziente da minimizzare un nistagmo spontaneo attraverso la fissazione visiva ed ingrandiscono gli occhi del paziente rendendo più facile raccogliere i movimenti degli occhi sottili. I pazienti devono essere testati con i propri occhiali o contatti in atto per ottimizzare la loro visione. Nella popolazione pediatrica, è richiesto il giudizio, come non tutti i bambini tollerano il suo utilizzo, che è una delle ragioni per riservare tale parte dell'esame per ultimo. L'esame neurologico è costituito da una serie di test di funzionalità cerebellari, come finger-to-nose test, test tallone-tibia, movimenti alternati rapidi, il test Romberg, e l'andatura.
Nel test dito-naso, il paziente viene chiesto di alternare toccare il dito dell'esaminatore estesa (tenuto su condizioni di mercato da parte del paziente) e la sua / suo proprio naso alternativamente il più velocemente e accuratamente possibile. Questo viene fatto prima con occhi aperti e poi con gli occhi chiusi. Se il paziente overshoot il bersaglio in modo coerente, la malattia cerebellare è sospetto. Questi pazienti possono anche a volte presentare un tremito, come il dito si avvicina al bersaglio. ;
A prima vista;
Esame neurologico
• Valutazione dei movimenti muscolari oculari
• Prova dito-naso
• Prova tallone-tibia
• movimenti alternati rapidi
• test di Romberg
• Valutazione della deambulazione
Durante il test del tallone-tibia , si dovrebbero osservare movimenti fluidi Con la malattia cerebellare, diventa evidente una oscillazione, nel movimento tallone-tibia .
I Movimenti alternati rapidi sono testati in due modi. Al paziente viene chiesto di toccare alternativamente la coscia con il palmo ed il dorso della loro mano in rapida successione, o il paziente è invitato a toccare il pollice e ciascuna delle dita in mano rapidamente avanti e indietro. La possibilità di eseguire un rapido movimento alternato è l’adiadococinesia; l'incapacità di farlo è chiamato dis adiadococinesia ed è indicativo della patologia cerebellare.
Con la locuzione Test di Romberg si intende un particolare esame diagnostico comunemente adoperato in Neurologia e Otorinolaringoiatria su pazienti che lamentano disordini dell'equilibrio o instabilità (atassia). La prova fu ideata dal neurologo Moritz Heinrich Romberg (1795-1873).
 La
procedura è semplice: il medico chiede al paziente di stare in piedi a talloni
uniti e braccia distese in avanti per un tempo di alcuni secondi ad occhi
aperti. Si fa ripetere l'esame al paziente chiedendogli di chiudere gli occhi.
Se tendesse a barcollare fortemente o cadere nei primi 30 secondi, il test si
intende positivo in caso di ATASSIA DI INFORMAZIONE (presenza di deficit di
informazione sensitiva propriocettiva e labirintica), mentre è negativo in caso
di ATASSIA CEREBELLARE. Una lieve oscillazione non è da considerarsi
patologica. Non è infrequente il cosiddetto falso positivo, ossia la perdita di
equilibrio in pazienti sani ma affetti da disturbi psicologici (ansia, stress).
In tali casi il medico, che riconosce facilmente un soggetto ansioso, propone,
durante il test, piccoli diversivi o distrazioni, come tracciare dei segni col
dito sulla fronte del paziente, oppure fargli tastare coi pollici le altre 4
dita.
La
procedura è semplice: il medico chiede al paziente di stare in piedi a talloni
uniti e braccia distese in avanti per un tempo di alcuni secondi ad occhi
aperti. Si fa ripetere l'esame al paziente chiedendogli di chiudere gli occhi.
Se tendesse a barcollare fortemente o cadere nei primi 30 secondi, il test si
intende positivo in caso di ATASSIA DI INFORMAZIONE (presenza di deficit di
informazione sensitiva propriocettiva e labirintica), mentre è negativo in caso
di ATASSIA CEREBELLARE. Una lieve oscillazione non è da considerarsi
patologica. Non è infrequente il cosiddetto falso positivo, ossia la perdita di
equilibrio in pazienti sani ma affetti da disturbi psicologici (ansia, stress).
In tali casi il medico, che riconosce facilmente un soggetto ansioso, propone,
durante il test, piccoli diversivi o distrazioni, come tracciare dei segni col
dito sulla fronte del paziente, oppure fargli tastare coi pollici le altre 4
dita.
Il sintomo: vertigine
La sensazione di vertigine ha origine da una distorsione della sensazione di movimento del corpo nello spazio. Questa distorsione può essere una rotazione (come una giostra) oppure una traslazione (sensazione di caduta di spinta, di basculamento) o semplicemente una sensazione d'instabilità.
Il bambino molto piccolo che non può esprimere ciò che sente si aggrappa ai genitori, chiede di essere portato, rifiuta di mettersi in piedi e si addormenta. Quando il bambino si può esprimere può dire: «la casa gira». Il bambino un poco più grande non ha sempre spontaneamente un vocabolario sufficiente per definire la sua sensazione e si soddisfa molto rapidamente della parola vertigine immediatamente proposta da coloro che lo circondano. Durante l'anamnesi è importante proporre diverse definizioni a un bambino che ha dei disturbi dell'equilibrio, mimarle affinché egli scelga quella che corrisponde maggiormente a ciò che avverte e avvicinarsi così alla diagnosi.
I disturbi dell'equilibrio nel bambino possono tradursi con delle cadute frequenti, un'impossibilità a tenersi in piedi (atassia) o disturbi della deambulazione.
La sensazione di vertigine può essere molto ben sopportata o al contrario associata a nausea, inappetenza, vomito, dolori addominali («mal di pancia»), cefalee e disturbi neurovegetativi (pallore, tachicardia che può arrivare fino al malessere vagale).
Donde vengono le sensazioni di movimento e la sensazione vertiginosa?
Noi percepiamo il movimento della nostra testa grazie a tre tipi di recettori di movimento: i recettori vestibolari, visivi e somestesici (superficiali e profondi o propriocettivi). I recettori vestibolari sono situati nell'orecchio interno, cavità ossea scavata nella rocca, che ospita anche l'apparato dell'udito. Essi percepiscono i movimenti di rotazione e di traslazione nonché la posizione della testa rispetto alla gravità. I recettori visivi percepiscono i movimenti del nostro corpo nello spazio rispetto a punti di riferimento vicini o lontani. I recettori propriocettivi situati a livello dei tendini, delle articolazioni, della cute percepiscono i movimenti e la posizione delle differenti parti del nostro corpo nonché il loro contatto al suolo (per esempio pianta dei piedi durante la marcia, natiche in posizione seduta) (Figura 1).
Punto essenziale
La distorsione percettiva responsabile delle vertigini può avere origine dal cattivo funzionamento di uno o più di questi recettori sensoriali di movimento, ma anche di tutte le strutture centrali partecipano all'analisi e all'integrazione delle informazioni di movimento e di posizione del capo e del corpo.
Disfunzione di uno o più recettori sensoriali di movimento
La sensazione di vertigine può avere origine in primo luogo dal vestibolo e/o dagli occhi e più accessoriamente dai recettori propriocettivi. La disfunzione vestibolare può provocare una sensazione vertiginosa (grande vertigine rotatoria, lateropulsione, caduta, beccheggio) che può essere breve o di diverse ore, legata ai movimenti o aggravata dai movimenti della testa. Vi si associano spesso in fase acuta dei movimenti caratteristici degli occhi: il nistagmo. Il nistagmo è composto da movimenti coniugati dei due occhi che associano una fase lenta seguita da una fase rapida. La fase rapida è importante da riconoscere in clinica perché indica il lato dei recettori vestibolari più attivi (una lesione destra si accompagna per esempio a un nistagmo sinistro di direzione costante). Anche un disturbo visivo può essere responsabile di vertigini , specialmente quando giunge ad alterare la visione binoculare dinamica (visione asimmetrica, disturbi di vergenza). Le vertigini sono allora spesso legate all'eccessivo lavoro visivo (lunghe sedute di videogiochi, al computer, alla televisione, di lettura), comparendo ai movimenti della testa o del solo sguardo, ma una causa visiva non provocherà mai da sola una grande vertigine rotatoria di varie ore, a differenza di un disturbo vestibolare. I disturbi della propriocezione non causano grandi vertigini, ma sensazioni di instabilità o cadute frequenti.
Disfunzione delle strutture centrali che partecipano all'analisi e all'integrazione
dei movimenti e delle posizioni del corpo
Dopo i recettori sensoriali, l'informazione di movimento è integrata a livello del tronco cerebrale e quindi «interpretata» da un numero molto elevato di strutture cerebrali, coinvolte in funzioni cognitive e affettive. Una disfunzione a uno dei livelli di integrazione può provocare una distorsione percettiva. In questo caso la risposta dei recettori può essere normale, legata a una stimolazione periferica reale, ma la sua interpretazione centrale è errata o eccessiva (per esempio cinetosi), oppure la risposta dei recettori è indipendente da ogni stimolazione periferica e generata dai centri stessi (l'esempio tipo di una sensazione vertiginosa puramente centrale è la vertigine da altezza).
Esame clinico oto-neuro-vestibolare del bambino con vertigini
Al letto del malato è già possibile realizzare un rapido esame clinico otologico vestibolare e neurologico.
La sindrome vestibolare si può riconoscere da una perdita di equilibrio lateralizzata: tendenza alla caduta sempre dallo stesso lato a occhi chiusi e in piedi su un terreno duro o sul materasso, il che altera le informazioni propriocettive e sensibilizza il test.
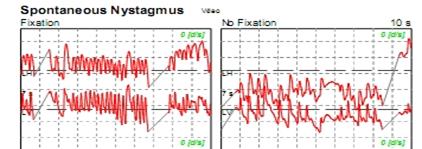
L’osservazione degli occhi può rivelare un battito regolare (alternante fasi rapide e fasi lente) che corrisponde a un nistagmo. Questo nistagmo è rapidamente inibito dalla fissazione oculare e quindi evidente solo grazie a degli occhiali che rendono sfocata la visione (occhiali di Frenzel). La fase rapida del nistagmo indica il vestibolo più eccitabile (e sarà di direzione opposta a una lesione). Si ricerca un nistagmo provocato dalle posizioni coricate sui lati (centrale o periferico).
Bisogna verificare se il bambino sente bene dalle due orecchie: scuotendo presso l’orecchio un oggetto sonoro (per esempio delle chiavi) senza che il bambino lo veda, sussurrando a un orecchio delle parole che deve ripetere mentre si tappa il padiglione dell’altro orecchio muovendo il trago per realizzare un mascheramento e impedire a questo orecchio di rispondere al posto dell’altro. Questa acumetria clinica si può realizzare chiedendo al bambino di indicare immagini o disegni che gli si mostra su carta e che corrispondono alle parole sussurrate a 30 cm dal suo orecchio.
Bisogna praticare un’otoscopia e un esame neurologico veloce ma completo.
I primi esami diagnostici da richiedere al minimo dubbio sono l’esame audiovestibolare e il controllo oftalmologico.
il primo e più importante passo nella valutazione è ottenere una storia completa. Bisogna prima stabilire la presenza di vertigine contro qualche altra forma di vertigini da luogo ad una storia della sensazione di rotazione o movimento. Questo può risultare difficile nei bambini più piccoli, dove può essere utile per evocare i sinonimi parco giochi come la filatura, oscillare, come un-giostra ecc
Al letto del malato è già possibile realizzare un rapido esame clinico otologico vestibolare e neurologico.
La sindrome vestibolare si può riconoscere da una perdita di equilibrio lateralizzata: tendenza alla caduta sempre dallo stesso lato a occhi chiusi e in piedi su un terreno duro o sul materasso, il che altera le informazioni propriocettive e sensibilizza il test.
L'osservazione degli occhi può rivelare un battito regolare (alternante fasi rapide e fasi lente) che corrisponde a un nistagmo. Questo nistagmo è rapidamente inibito dalla fissazione oculare e quindi evidente solo grazie a degli occhiali che rendono sfocata la visione (occhiali di Frenzel). La fase rapida del nistagmo indica il vestibolo più eccitabile (e sarà di direzione opposta a una lesione). Si ricerca un nistagmo provocato dalle posizioni coricate sui lati (centrale o periferico).
Bisogna verificare se il bambino sente bene dalle due orecchie: scuotendo presso l'orecchio un oggetto sonoro (per esempio delle chiavi) senza che il bambino lo veda, sussurrando a un orecchio delle parole che deve ripetere mentre si tappa il padiglione dell'altro orecchio muovendo il trago per realizzare un mascheramento e impedire a questo orecchio di rispondere al posto dell'altro. Questa acumetria clinica si può realizzare chiedendo al bambino di indicare immagini o disegni che gli si mostra su carta e che corrispondono alle parole sussurrate a 30 cm dal suo orecchio.
Bisogna praticare un'otoscopia e un esame neurologico veloce ma completo.
I primi esami diagnostici da richiedere al minimo dubbio sono l'esame audiovestibolare e il controllo oftalmologico.
Esame vestibolare clinico di base
L'esame audiovestibolare viene realizzato da un otorinolaringoiatra e comprende un'anamnesi «poliziesca»dei familiari e del bambino se questo è abbastanza grande da raccontare ciò che gli è accaduto, e una serie di test clinici.
Questi test clinici sono accessibili senza alcun materiale sofisticato, cioè a disposizione di ogni medico. Li tratteremo con in modo discretamente dettagliato.
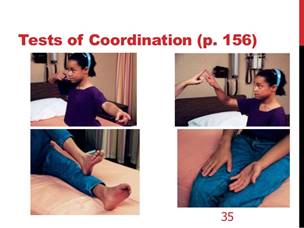
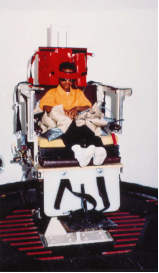
• Osservazione del comportamento posturale e motorio spontaneo durante i giochi, studio del controllo posturale durante la deambulazione, il salto su terreno duro e su terreno molle, se possibile con gli occhi chiusi (Figura 2, Figura 3 ). Un deficit vestibolare bilaterale si accompagna a cadute frequenti e a squilibrio in occasione di rotazioni rapide della testa e, nel bambino molto piccolo, a un'ipotonia assiale.
|
|
|
|
Figura 2. A,B Ricerca di una deviazione posturale statica e dinamica. Su terreno duro o terreno molle, occhi aperti o chiusi (A,B,C). |
Figura 2.C Romberg occhi chiusi |
|
|
|
|
|
|
|
|
|
Figura 3 : Esame dell'attività spontanea (da A a D). Wiener-Vacher S.R.,ET AL.,1997/8/2001.
|
|
|




• Studio dell'oculomotricità (Figura 4 ): esso comporta lo studio dei movimenti coniugati degli occhi durante l'inseguimento di un piccolo bersaglio sul piano frontale e sul piano sagittale mediano (per la convergenza oculare). Esso comporta anche lo studio della precisione dei movimenti saccadici oculari quando si chiede al bambino per esempio di guardare un piccolo giocattolo che compare dietro un cartone perforato. Figura 4 :Aa gaze nistagmus
Studio dell'oculomotricità.
|
|
|
|
|
|
|
Fig. 4A gaze nystagmus |
4B inseguimento mobilità e convergenza oculare |
4C otticocinetica |
D saccadi |
D saccadi |




Figura 5a. Ricerca di un nistagmo spontaneo o rivelato da scuotimento della testa. Esame
sotto occhiali di Frenzel o videoscopia per evitare l’inibizione di un eventuale nistagmo dovuta alla fissazione. Manovra di scuotimento rapido della testa per rivelare una preponderanza direzionale (che può essere periferica o centrale).
|
|
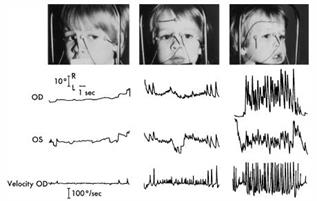
Figura 5b Nistagmo registrato con ENG con gli occhi ruotati a sinistra . Questo paziente aveva la testa girata spontaneamente verso destra . ( Da Noorden GK von , Munoz M , Wong SY : . Meccanismi di compensazione a nistagmo congenito Am J Ophthalmol 104 : 387 , 1987 )
Le seguenti tre tracce sono dI un paziente con CN(Nistagmo Congenito) a causa di un disturbo visivo sin dai primi anni di vita.
|
|
|
Figura 5c CN: è tipico un nistagmo molto più intenso con la luce (fissazione) rispetto al buio. Questo è il modo più semplice per diagnosticare un NC rispetto ad ogni altro tipo di nistagmo. |
|
Figura 5d CN: l’inseguimento orizzontale è interrotta da una forte nistagmo |
Ricerca di saccadi di richiamo durante impulsi rotatori rapidi della testa (nel piano orizzontale e nel piano sagittale) mentre il soggetto fissa lo sguardo su un bersaglio (head impulsion test di Halmagyi) (Figura 6 ). La presenza di una saccade di richiamo indica un deficit completo del canale semicircolare situato nel piano di rotazione e sul lato della rotazione. Questa manovra può essere eseguita nel piano dei 6 canali semicircolari. Tuttavia si può ottenere una saccade di richiamo quando esiste un difetto di convergenza oculare o una ambliopia, in questo caso la saccade ottenuta è meno riproducibile e in caso di disturbi di convergenza oculare scompare quando il bersaglio è posto a distanza (più di 1,50 m).
|
Figura 6. . Test di impulso rotatorio della testa HEAD THRUST TEST O TEST DI HAMALGYI Si invita il bambino a fissare un bersaglio posto al centro del campo visivo (ad esempio, il naso dell’esaminatore), quindi si ruota bruscamente il capo in una direzione con un movimento veloce di circa 30°. Nel bambino normale l’occhio rimarrà sul bersaglio. Nella rotazione verso destra, infatti, il canale semicircolare laterale di destra comanderà una controrotazione dei bulbi verso sinistra che permetterà al paziente di non perdere la mira; se al contrario il riflesso vestibolo-oculomotore risulta deficitario, il movimento verso sinistra sarà insufficiente e gli occhi, per riprendere il bersaglio, dovranno compiere un movimento rapido verso sinistra .Il test di Halmagyi: può valutare i sei canali (A, B). Rotazione della testa passiva, rapida, breve, imprevedibile nel piano del canale studiato. Saccade di recupero: deficit vestibolare completo dal lato della rotazione della testa con saccade. Se disturbo di convergenza oculare: normale quando il bersaglio è allontanato.
|
|
|
|
|

|
HEAD SHAKING TEST L’esaminatore ruota il capo del paziente sul piano orizzontale per circa per 20 volte ad una frequenza di circa 2 Hz. Osserva quindi attraverso gli occhiali di Frenzel la comparsa di un eventuale nistagmo. Nei deficit vestibolari periferici compare un nistagmo diretto verso il lato sano. L’head shaking test non è un test molto sensibile, specìalmente se eseguito a distanza dalla lesione, ma è accettato come test di screening per evidenziare asìmmetrie vestibolari a livello periferico o centrale senza possibilità di precisazioni topodiagnostiche. Non deve essere l’unico test con cuì fare diagnosi di deficit periferico (IFigura 7). |
|
Figura
|

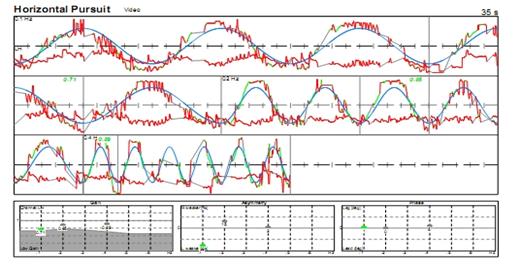
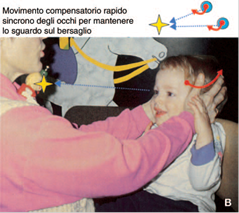
· Inibizione mediante la fissazione delle risposte vestibolo-oculari (VOR) e optocinetiche (OKN) (Fig. 8): il bambino seduto sulle ginocchia di un genitore o su una poltrona girevole deve mantenere l’attenzione e lo sguardo su un piccolo giocattolo o una caramella tenuti davanti a lui e che girano insieme a lui. Un bambino fin dall’età di 1 anno può mantenere fisso lo sguardo su un bersaglio e inibire al 100% i movimenti OKN e VOR che il movimento della poltrona provoca se è completamente attento durante la rotazione.
|
|
Figura 8a :Optocinetica e risposte vestibolo-oculari e loro inibizione mediante la fissazione. Inibizione al 100% nei bambini molto piccoli se l'attenzione è sostenuta. Se non esiste inibizione al 100%, età superiore ai 2 anni: lesione cerebellare.
|
|
|
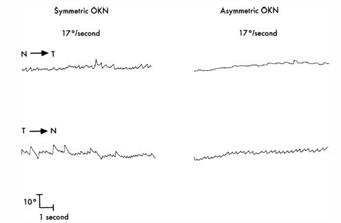
Figura 8b Nistagmogramma binoculare durante la stimolazione optocinetico nasotemporale ( tracciati superiori) e temporonasale ( tracciati inferiori) in un soggetto normale ( a sinistra) e un paziente con esotropia essenziale infantile essenziale ( a destra ) . Si nota scarsa risposta optocinetica quando il tamburo è mosso in una direzione nasotemporale nel paziente esotropico . ( Da Noorden GK von : concetti attuali di esotropia infantile essenziale ( Bowman Lecture ) Eye 2 : . 343 , 1988 )
|

|
|
|
Figura 8d CN: OKN può essere assente (come in questo caso) o invertito. |
Ricerca di un nistagmo provocato dalla posizione di decubito laterale destro e sinistro (Figura 9 ). Si può porre il bambino in posizione distesa sul lato, su un lettino di visita o in braccio a un genitore. Lo sguardo del bambino è mantenuto indirizzato verso un giocattolo oppure un punto nella stanza. Si può mettere così in evidenza un nistagmo che evoca una canalolitiasi (vertigine posizionale parossistica benigna rara nel bambino) o un nistagmo di origine centrale.
|
|
Figura 9 : Ricerca di un nistagmo rivelato da cambiamenti di posizione. Vertigine posizionale parossistica benigna (cupulocanalolitiasi): latenza, geotropica, esauribile, adattabile, orizzonto-rotatorio, rara nel bambino. Lesione centrale: non latenza, non adattabile né esauribile, direzione variabile, multidirezionale, raramente isolato.
|
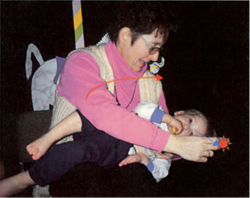
• Il test calorico (Figura 10 ) è un complemento indispensabile dell'esame clinico vestibolare (vedi sotto).
Esame neurologico clinico
È indispensabile davanti a un paziente portatore di vertigini o di disturbi dell'equilibrio. Può essere effettuato molto rapidamente (meno di 10 min) e se si prende l'abitudine di eseguire questo esame, sarà tanto più facile riconoscere un'anomalia quando sarà presente.
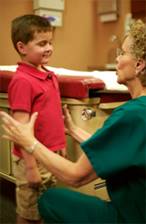
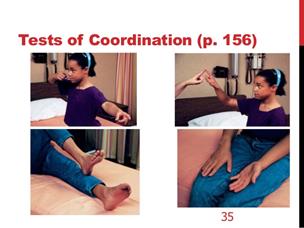
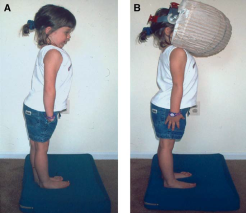
L'esame neurologico è costituito da una serie di test di coordinazione /funzionalità cerebellari , come il test indice naso, il test tallone- tibia , movimenti alternati rapidi , il test Romberg , e la marcia
Nel test indice - naso , il paziente viene chiesto di toccare alternativamente il dito dell'esaminatore estesa ed il suo proprio naso alternativamente il più velocemente e accuratamente possibile . Questo viene fatto prima con occhi aperti e poi con occhi causati se il paziente sbaglia il bersaglio in modo coerente , la malattia cerebellare è sospetto. Questi pazienti possono anche a volte presentare un tremito ,quando il dito si avvicina al bersaglio .Il Test di Romberg può essere meglio studiato con la Pedana Stabilometrica Statica e Computerizzata
|
|
|
|
Figura 10 di test di coordinazione |
il test Romberg |
|
|
|
|
Pedana Stabilometrica Statica |
Pedana Stabilometrica Computerizzata |
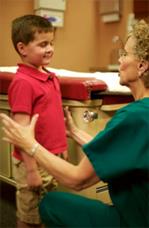
Esame delle coppie craniche
• L'olfatto (I): il compito sarà di determinare qual è tra le nostre due mani quella che è stata profumata con una goccia di vaniglia.
• La vista (II) sarà valutata con l'inseguimento binoculare quindi monoculare (l'altro occhio resta coperto), il compito sarà seguire un piccolo giocattolo, e il campo visivo (il bambino fissa il nostro naso e deve afferrare le dita delle nostre mani che si muovono e non quelle che restano immobili nelle zone molto laterali del campo visivo).
• L'oculomotricità (III, IV, VI), testata fin dall'inizio dell'esame vestibolare, valuta lo spostamento coniugato dei due occhi durante il compito di inseguimento di un piccolo giocattolo che si sposta nel piano frontale quindi che si avvicina verso la radice del naso, per giudicare la qualità e la simmetria della convergenza oculare; il III paio cranico viene anche valutato per la sua parte intrinseca con la contrazione pupillare ottenuta attraverso l'illuminazione della pupilla con la luce dell'otoscopio (dallo stesso lato o consensualmente).
• La sensibilità del volto (V) sarà valutata mediante palpazione ma anche con la sensazione fredda percepita al contatto con la superficie metallica dell'abbassalingua sulla faccia iugale delle guance e la cute del volto.
• La motricità del volto durante la realizzazione di smorfie (fischiare, soffiare, muovere il naso, sbarrare gli occhi) e soprattutto la simmetria delle contrazioni dei muscoli del viso attestano il buon funzionamento del nervo faciale (VII).
• La contrazione simmetrica del velo e della faringe con l'abbassalingua nonché l'assenza di false strade durante l'ingestione di bevande testimoniano l'integrità dei nervi misti (IX, X, XI) e la simmetria di forza di sollevamento delle spalle quella del XI spinale.
• La simmetria della lingua quando il bambino la espone e la sposta a destra e a sinistra esclude una lesione del XII.
Esame neurologico somatico clinico (Figura 11)
Figura 11 :
Esame neurologico: paia di nervi cranici, tono, forza muscolare, sensibilità, riflessi osteotendinei, ricerca del segno di Babinski, di segni di lesione cerebellare.
A. Segno del fazzoletto.
B. Tono degli arti inferiori.
C. Riflesso osteotendineo.
Si verifica la somestesia (caldo-freddo, puntura-tocco), la motricità, il tono muscolare (ricercando una spasticità o un'ipotonia confrontando i lati destro e sinistro); si ricerca la presenza dei riflessi osteotendinei ai quattro arti, il segno di Babinski (anormale estensione delle dita in occasione della stimolazione del bordo laterale della pianta dei piedi) e segni di lesione cerebellare (riflessi osteotendinei [ROT] vivaci diffusi policinetici, ruota dentata, dismetria nella prova dito-naso o tallone-ginocchio o grande imprecisione dei gesti in occasione di compiti di prensione di piccoli giocattoli per porli in una piccola ciotola, adiadococinesia durante i movimenti di marionette con le mani), nonché dei segni che possono venire ad aggiungersi a un'ipermetria dei movimenti saccadici oculari (l'occhio supera il bersaglio), un inseguimento oculare saccadico o un difetto di inibizione mediante la fissazione oculare dei riflessi optocinetico e vestibolo-oculare già riscontrato durante lo studio dell'oculomotricità, e che completeranno la sindrome cerebellare
Figura . Esame neurologico: paia di nervi cranici, tono, forza muscolare, sensibilità, riflessi osteotendinei, ricerca del segno di Babinski, di segni di lesione cerebellare.
A. Segno del fazzoletto.
B. Tono degli arti inferiori.
C. Riflesso osteotendineo.
D. Fascicolazione alla flessione del piede.
E. 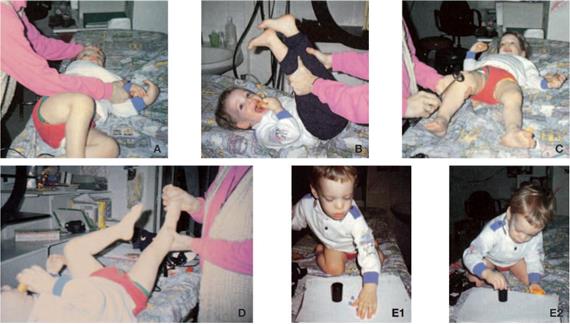 Precisione dei
gesti fini.
Precisione dei
gesti fini.
Lo studio della sensibilità profonda può essere eseguito agli arti superiori: si chiede al bambino, mentre tiene gli occhi chiusi, di afferrare con una delle mani il pollice dell'altra mano che è portata in una posizione qualsiasi dall'esaminatore (se non esiste un deficit motorio, un interessamento della sensibilità profonda si tradurrà in un atto mancato, con la mano che non potrà essere trovata a occhi chiusi dal lato del deficit). Lo stesso esame può essere eseguito agli arti inferiori con il movimento talloni-ginocchia a occhi chiusi o la determinazione a occhi chiusi della posizione impressa alle dita dei piedi (in alto o in basso) dall'esaminatore.
Nel bambino piccolo (fino a 4-5 anni), l'esame confronta i due emisomi: durante la motricità spontanea, durante i giochi di prensione di oggetti o dell'aggrapparsi a un appoggio con le mani al momento del passaggio dalla posizione supina a quella in piedi, durante la deambulazione o la corsa. La tonicità muscolare è confrontata durante flessioni ed estensioni agli arti superiori (segno del fazzoletto), agli arti inferiori (in flessione ed estensione a gambe flesse e gambe tese) e a livello del dorso (flessione ed estensione della schiena). I ROT sono valutati ai quattro arti. La ricerca del riflesso cutaneo-plantare permette di individuare un segno di Babinski (alla stimolazione della pianta del piede, si ottiene in teoria una flessione delle dita appena la deambulazione autonoma è stata acquisita; il segno di Babinski è un'estensione delle dita: è fisiologico prima che sia acquisita la deambulazione autonoma).
Il test dell'udito è complementare in caso di vertigini .
Una vertigine o un disturbo dell'equilibrio possono essere il segno di una lesione dell'orecchio interno, dove si trovano gli apparati dell'udito e dell'equilibrio. I test dell'udito sono indispensabili davanti ad ogni bambino affetto da vertigini (o instabile) a complemento dell'esame vestibolare e neurologico per valutare la funzione delle due modalità funzionali dell'orecchio interno.
L'udito sarà valutato con un audiogramma tonale e vocale. Possono essere effettuati test più completi in centri specializzati di esplorazione funzionale audio-vestibolo-oculare (otoemissioni, riflessi di orientamento condizionati [ROC], potenziali evocati uditivi [PEA]), che però possono essere realizzati in un secondo tempo.
Esame oftalmologico
Esso deve comprendere una misurazione dell'acuità visiva, ma anche una misurazione della rifrazione oculare sotto dilatatori del sistema di accomodazione (tipo Ciclolux® o Tropimil®) senza dimenticare una valutazione ortottica. Orbene, pochi oftalmologi effettuano essi stessi una valutazione ortottica, compito che in Francia è generalmente svolto da ortottisti. Questo esame statico valuta i limiti di fusione della visione binoculare con l'aiuto di prismi che obbligano l'occhio a divergere o convergere per fondere le immagini delle due retine.
Ciò è realizzato per la visione da vicino e quella da lontano. Questo esame permette di diagnosticare esoforie (differenza di altezza degli occhi), strabismi o difetti di convergenza oculare. Non esiste ancora un test riassuntivo valido che permetta una valutazione obiettiva delle capacità di vergenza oculare dinamica (studio in corso).
I metodi di valutazione della funzione vestibolare hanno fatto grandi progressi da una decina d'anni e abbiamo ora la possibilità di realizzare una valutazione molto completa della funzione vestibolare canalare e otolitica. Il test di base è tuttavia più accessibile per ogni otorinolaringoiatra è il test calorico che, benché molto incompleto (e bisogna conoscerne i limiti), permette di distinguere nella grande maggioranza dei casi le malattie vestibolari da quelle che non lo sono e di individuare i disturbi periferici anche antichi e ben compensati. Questo test calorico è quindi in primo piano nell'esame vestibolare di base.
La valutazione della funzione vestibolare si realizza registrando le risposte vestibolo-oculari e vestibolo-spinali ottenute in risposta a stimolazioni vestibolari. Le stimolazioni vestibolari possono interessare il sistema dei canali semi-circolari (è il caso del test calorico o delle rotazioni secondo un asse verticale) o gli organi otolitici (è il test OVAR oppure RAIG, i potenziali miogeni sacculo-collici, la misurazione della verticale soggettiva).
Test della funzione canalare
Il sistema vestibolare funziona in un ampio intervallo di frequenze. Pertanto, ha senso intuitivo e clinico che per testare il maggior numero di queste frequenze come possibili (per replicare il mondo reale) prima di effettuare un piano di diagnosi e riabilitazione per un bambino con insufficienza vestibolare. Per esempio, il test calorico è sensibile alle basse frequenze (0,004 Hz)
Test calorico (Figura 12)
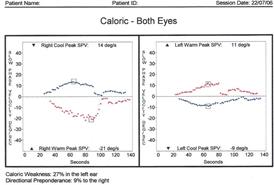
Si utilizza l'irrigazione di ogni condotto uditivo esterno successivamente con acqua a 30° C e 44° C per 30 secondi, per provocare un movimento dell'endolinfa dei canali semicircolari esterni posti in posizione verticale, movimento che indurrà delle scosse coniugate degli occhi (chiamate nistagmo) che corrispondono alla risposta vestibolo-oculare. I canali orizzontali sono portati in posizione verticale installando il bambino su un piano inclinato di 30° rispetto alla linea orizzontale. Il numero di nistagmo è misurato durante 30 secondi a partire dal 30o secondo dopo l'inizio dell'irrigazione, eseguendo un confronto tra i differenti stimoli. Questi risultati sono rappresentati su un diagramma messo a punto da G. Freyss e che visualizza la simmetria delle risposte dei due orecchi destro e sinistro (o valenza) e l'equilibrio dei sistemi nistagmici destro e sinistro (detto preponderanza direzionale normalmente < 15%). Appena dopo l'irrigazione e la misurazione della risposta si può posizionare il bambino in piedi, se possibile con gli occhi chiusi, per vedere se la stimolazione del vestibolo è responsabile di una deviazione posturale (a 30 ° C, caduta verso il lato stimolato, a 44° C, caduta verso il lato opposto). Una tale deviazione, quando la si osserva, è indice del buon funzionamento della via vestibolospinale dal lato stimolato. Per ridurre il tempo associato con il test calorico (cioè, per renderlo più "amico dei bambini"), si consiglia di utilizzare un test di screening caldo monotermico (MWST). MWST offre una buona sensibilità e specificità (83% e 90%) rispetto al test calorico bitermico a bambini dai 6 ai 14 anni.noi non utilizziamo più la stimolazione con acqua ,ma ad aria vedi immagine 12c e 12d
|
|
|
|
|
|


|
Figura 12. Test calorico: indispensabile Fig. 12 A :Irrigazione con acqua a 30° C e 44° c/30 sec ad ogni orecchio. Esame della risposta vestibolo-oculare (Frenzel o video) con una misurazione della frequenza delle fasi rapide o delle velocità delle fasi lente. Fig. 12 B :Diagramma di G. Freyss che mostra qui una ipovalenza sinitra senza preponderanza direzionale. Fig. 12 C Stimolatore Ad Aria Otometrics da noi utilizzata Fig. 12 D Chartr 200 VNG/ENG |
Esame pendolare (Figura 11A,B)
Il soggetto è seduto su una sedia girevole e sottoposto a rotazioni sinusoidali (di frequenza 0,05 Hz e di velocità di 25° /s). Il riflesso vestibolo-oculare registrato corrisponde alla stimolazione dei canali semicircolari esterni e la frequenza e la velocità delle fasi lente del nistagmo devono essere normalmente simmetriche. Le asimmetrie sono presenti in fase acuta dopo una lesione periferica del vestibolo e sono molto rapidamente cancellate dai processi di compensazione centrale. Un'areflessia bilaterale si presenta come un'assenza di risposta.
PRINCIPALI PROVE PENDOLARI
1e)Prova pendolare
La
prova pendolare utilizza uno stimolo acceleratorio di tipo sinusoidale, di cui
sono noti l’ampiezza ed il periodo dell’oscillazione.Tale prova si differenzia
dalle precedenti in quanto lo stimolo acceleratorio
viene applicato alternativamente ai due labirinti: tale variazione continua
(oraria ed antioraria) di stimolazione crea una situazione che si avvicina alla
riproduzione dei naturali movimenti del capo durante la vita di relazione.
Lo stimolo rotatorio sinusoidale è definito da due variabili semplici:
a) il periodo d’oscillazione,
b) l’ampiezza d’oscillazione.
Con la prova pendolare la sedia deviata dalla sua posizione d’equilibrio ritorna a questa posizione con un’oscillazione sinusoidale smorzata; lo stimolo fa deviare la cupola alternativamente in direzione ampullipeta e ampullifuga, producendo un nistagmo che cambia direzione in ogni emiciclo rotatorio (periodo 20 sec., ampiezza massima di partenza 180°, smorzamento in 15 periodi) la velocità angolare della fase lenta (VAL) massima o media e la frequenza massima sono usate per quantificare la risposta in ogni emiciclo.
Rotazioni orizzontali sinusoidali
1e)Test pendolare rotatorio sinusoidale smorzato (Grenier)
Esso presenta le seguenti caratteristiche: un periodo di 20 secondi (frequenza 0,05 Hz), un'ampiezza massima di 180°, uno smorzamento esponenziale in 15 periodi, un'accelerazione in partenza di 18°/s2. Il movimento oculare è registrato contemporaneamente al movimento della poltrona. Il parametro studiato è il più delle volte la velocità media della fase lenta del nistagmo, calcolata mediante computer, o l'ampiezza cumulativa del movimento dell'occhio. Quest'ultima è ottenuta addizionando le fasi lente e sopprimendo le fasi rapide. Il tracciato cumulativo ha allora la forma di un sinusoide smorzato sovrapponibile al movimento della poltrona. Sono allora misurati il guadagno e la fase del RVOH. Nel soggetto normale, essi sono rispettivamente vicini a 0,6 per il guadagno e a 10° per la fase. Questo test ha il vantaggio di essere rapido, ma esamina il sistema vestibolare soltanto nel campo delle risposte a basse frequenze, mentre può rispondere a un'ampia gamma di frequenze che vanno da 0,01 Hz a 20 Hz.
Si studia anche nel corso di questo test l'indice di inibizione del nistagmo con fissazione oculare (IFO). In questo caso, si chiede al soggetto di fissare un bersaglio luminoso posto nella maschera di videonistagmografia mentre è sottoposto a questa rotazione. La fissazione oculare induce un'inibizione di oltre il 50% del guadagno del RVOH. Un IFO superiore al 50% è sempre segno di una patologia vestibolare centrale, ma non ha un valore localizzatorio preciso.
In caso di distruzione acuta e unilaterale di un labirinto, si osserva una diminuzione bilaterale del guadagno del RVOH a questo test a basse frequenze, più marcata per le rotazioni verso il lato leso che per quelle verso il lato sano. A distanza, queste alterazioni a basse frequenze (0,05 Hz) scompaiono il più delle volte grazie al compenso vestibolare centrale il che, al contrario delle prove caloriche, limita l'interesse di questo esame quando è eseguito a distanza dalla lesione.
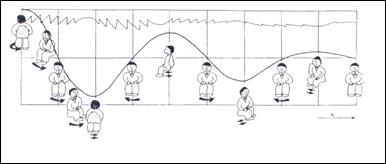
Fig.1-16
a velocità angolare
progressivamente
smorzata
2e)Test pendolare sinusoidale a scansione di frequenza. Ulmer E. 2002, Jenkins H.A., 1982 ;
Permette di analizzare la risposta del vestibolo su una più ampia gamma di frequenze. Nel corso di questo test, il paziente è collocato su di una sedia animata da un movimento sinusoidale il cui periodo passa progressivamente in 2 minuti da 20 a 2 secondi, il che corrisponde a uno spostamento della frequenza da 0,05 Hz a 0,5 Hz. L'ampiezza dell'oscillazione diminuisce progressivamente, in modo da mantenere un'accelerazione sensibilmente costante. Si può quindi valutare il guadagno di RVOH su una gamma di frequenze di stimolazione che vanno da 1 a 10. Un'areflessia alle prove caloriche e rotatorie sinusoidali può non verificarsi a questo test di scansione di frequenza. Ciò potrebbe corrispondere a una funzione canalare orizzontale residua e in particolare a una persistenza delle cellule fasiche dell'epitelio neurosensoriale, di cui si sa che sono stimolate a frequenze superiori a 0,1 Hz.,ha avuto una diffusione limitatissima in campo otoneurologico, anche se resta una tecnica interessante .
3e)Test ad accelerazione sinusoidale
Rappresenta un’evoluzione dal punto di vista tecnico in quanto eseguita mediante specifiche sedie guidate da un software in grado di avvicinare il più possibile le stimolazioni al range di normale attività del VOR. Esistono stimolazioni mono e multifrequenziali. I test rotatori ad accelerazione sinusoidale utilizzano in genere accelerazioni armoniche (SHA): Sinusoidal Harmonic Acceleration (Mathog, 1972), che è la prova roto-acceleratoria più utilizzata negli USA, in particolare le frequenze di oscillazione 0.01, 0.02, 0.04, 0.08, 0.16, 0.32 e 0.64 Hz, con velocità angolare massima di 50° / secondo per ciascuna frequenza.
Anche in questo caso la rotazione oraria induce l'eccitazione del labirinto destro e la contemporanea inibizione del sinistro e viceversa.
La risposta del VOR ai test rotatori viene descritta da tre parametri delle risposte a 5 stimoli (0,01, 0,02, 0,04, 0,08 e 0,16 Hz) ad una velocità massima di 50°/s (nel caso di sospetta areflessia labirintica bilaterale può essere portata fino a 100°/s:
a) Guadagno: rapporto fra la velocità massima degli occhi e la velocità massima della testa;
b) Ritardo (fase) in risposta allo stimolo: costante di tempo nell'accelerazione impulsiva (tempo, espresso in secondi, perché la velocità massima della fase lenta del ny declini al 37% del suo valore massimo) e angolo di fase nell'accelerazione sinusoidale (misura della relazione temporale fra la massima velocità degli occhi e quella della testa).
c) Simmetria: rapporto della velocità massima degli occhi nella rotazione vero destra e verso sinistra: (VAFLdx-VAFLsn) / (VAFLdx+VAFLsn) x100. Questo tipo di prova riveste un ruolo importante nel tentativo di ottenere una diagnosi topografica della lesione: infatti uno sfasamento tra la curva relativa all’andamento della VAFL e quella della velocità angolare della sedia interesserebbe tutte le frequenze nel caso di patologia centrale, mentre sarebbe limitato alle basse frequenze di stimolo nelle vestibolopatie periferiche; inoltre, nel tempo, a differenza della patologia centrale, in caso di lesione periferica le risposte tenderebbero a divenire più simmetriche (Probst e al., 1983).
I moderni test rotatori consentono di valutare, oltre allo studio del VOR, anche l'interazione fisiologica tra il riflesso vestibolo-oculare e il riflesso visuo-oculare. Questo è possibile facendo ruotare il paziente ad occhi aperti in un ambiente illuminato, mantenuto stazionario, costituito in genere dalle strisce verticali del tamburo otticocinetico (interazione sinergica fra i riflessi vestibolo e visuo-oculare: Vis-VOR) e, ancora, facendolo ruotare in maniera solidale con le strisce del tamburo otticocinetico oppure mentre fissa una mira luminosa attaccata alla sedia e posta di fronte a lui (interazione antagonista fra i riflessi vestibolo e visuo-oculare: VOR-Fix).
IL Test con sedia Rotatoria è sensibile alle basse di frequenze medie (0,01-1,28 al Hz).
IL Test con sedia Rotatoria (RCT)
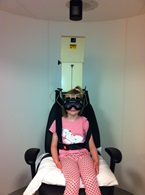 è una comune alternativa per il monitoraggio disturbi vestibolari progressivi
nei bambini. RCT viene utilizzato per monitorare disfunzione del sistema
vestibolare e compensazione nel tempo. Inoltre, RCT viene utilizzato per
valutare e monitorare la debolezza bilaterale vestibolare. In generale, i
protocolli RCT sono più comodo e più facilmente tollerato dai bambini, rispetto
ai test calorico tradizionali. In particolare, i bambini piccoli di solito
può sedersi in braccio a un genitore durante i test di rotazione, e il nistagmo
risultante è di solito meno intenso di quello che potrebbe essere stato
provocato tramite test calorico.
è una comune alternativa per il monitoraggio disturbi vestibolari progressivi
nei bambini. RCT viene utilizzato per monitorare disfunzione del sistema
vestibolare e compensazione nel tempo. Inoltre, RCT viene utilizzato per
valutare e monitorare la debolezza bilaterale vestibolare. In generale, i
protocolli RCT sono più comodo e più facilmente tollerato dai bambini, rispetto
ai test calorico tradizionali. In particolare, i bambini piccoli di solito
può sedersi in braccio a un genitore durante i test di rotazione, e il nistagmo
risultante è di solito meno intenso di quello che potrebbe essere stato
provocato tramite test calorico.
Quando viene utilizzato insieme con VNG, RCT completa la batteria di test per le valutazioni diagnostiche vestibolari. I vantaggi del RCT includono l'assenza di trasferimento di energia termica (che è di grande importanza per quanto riguarda la o test calorico bitermico), e valuta i RCT precisamente frequenze multiple basate su stimoli di rotazione (prove caloriche valutano solo le frequenze molto lente di circa 0,003 Hz). Fife et al 11 ha osservato in caso di sospetto e diagnosticato iporeflettività vestibolare bilaterale, RCT è preferita per i bambini di età compresa tra 3 e 10 anni.
Impulsi rotatori secondo un asse verticale (Figura 13 A, [4]
Il soggetto è seduto su una sedia girevole nell'oscurità. Questa sedia permette di applicare brevi accelerazioni e decelerazioni angolari secondo un asse verticale (40°/s2, per 1,5 s) le quali sono seguite da una velocità di rotazione costante. Questi impulsi rotatori producono rispettivamente un eccitamento e un'inibizione dei canali semicircolari esterni e una risposta vestibolo-oculare che verrà registrata e misurata (velocità delle fasi lente e durata della risposta). Questa risposta raggiunge molto rapidamente un massimo, quindi decresce progressivamente in 20-60 secondi in funzione dell'età del soggetto (meno di 18-20 sec nel bambino molto piccolo, più spesso 20 sec nell'adulto giovane). Un interessamento del vestibolo si manifesta con un'assenza di risposta dal lato colpito o con una diminuzione della risposta in ampiezza e in durata. Un'asimmetria delle risposte è segno di una lesione monolaterale. I processi di compensazione centrale cancelleranno progressivamente questa asimmetria, a spese tuttavia della durata delle risposte che sono allora molto brevi dai due lati (il che indica una grande probabilità di inibizione centrale).
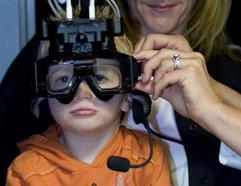
Test della rotazione intorno a un asse inclinato o test RAIG
Si tratta di uno studio della funzione otolitica stimolata da rotazioni a velocità costante intorno a un asse inclinato rispetto alla gravità, chiamato Raig o off vertical axis rotation (OVAR). È lo studio delle risposte nistagmiche otolitiche generali. Questa stimolazione vestibolare otolitica viene realizzata con una sedia rotatoria inclinabile guidata da un computer
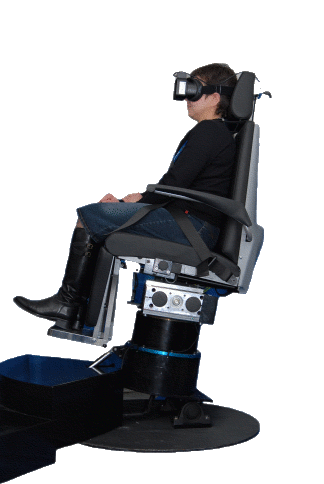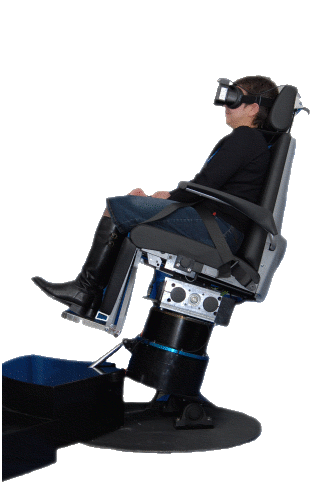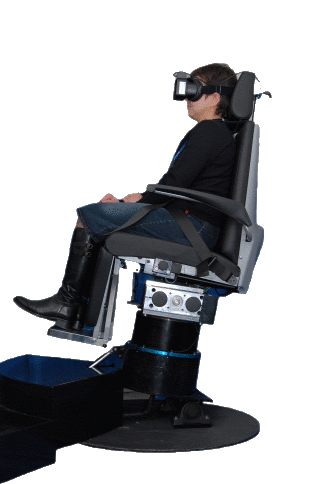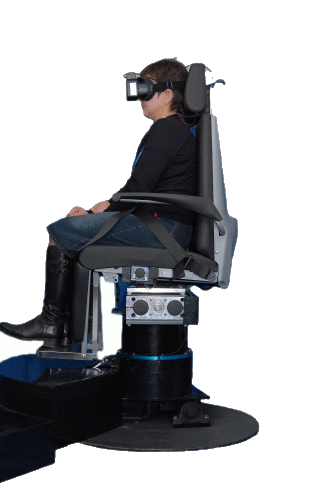

Fig.14a Questa Mega Torque è una sedia rotatoria con asse di rotazione fuori dall’asse verticale per il test degli Otoliti.
Fig.14b Il principale vantaggio clinico ,del test rotazionale informatizzato, è la capacità di produrre accelerazioni angolari che possono essere controllati e ripetuti . Stimoli multipli di varia intensità possono essere applicati al sistema vestibolare entro un tempo relativamente breve. Questi test permettono la valutazione del riflesso vestibolo-oculare e del percorsi neurale del sistema otticocinetico in un modo che non si ripetano procedure alternative.
Test OVAR o test RAIG: la poltrona girevole sulla quale il soggetto è seduto nell'oscurità è ruotata a velocità costante secondo un asse inclinato rispetto alla gravità. Questo movimento produce una stimolazione generale del sistema otolitico e in ritorno una risposta vestibolo-oculare che è registrata e misurata. Fig. 2-25 A,B
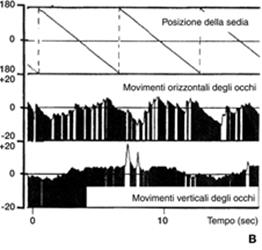
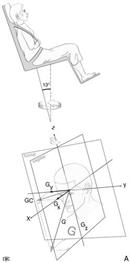
Fig. 15 A,B,C OVAR test (Off Vertical Axis Rotation: valuta la funzione otolitica) o test RAIG
A. La sedia è inclinata di 13° rispetto alla gravità e ruotata a una velocità costante di 60°/s (secondo Darlot C et al. Exp Brain Res 1988).
B. I due tracciati in basso mostrano le caratteristiche delle componenti orizzontali e verticali della risposta vestibolo-oculare otolitica in risposta a una rotazione OVAR oraria OVAR. La modulazione dei movimenti oculari (in posizione nonché in velocità delle fasi lente) è sincronizzata con la posizione della sedia durante la rotazione.
 La risposta vestibolo-oculare è registrata, al buio,
con elettronistagmografia o video-oculografia. La sedia Fig.
15 C
è sottoposta a un'accelerazione rotatoria breve, quindi a un movimento rotatorio
costante (60°/secondo) in base a un asse verticale; vengono quindi stimolati i
canali semicircolari orizzontali. A velocità di rotazione costante, la risposta
canalare si annulla progressivamente. La sedia è allora inclinata di 13°
rispetto alla gravità, sempre a velocità costante di 60°/secondo ; viene quindi
stimolato, in maniera elettiva, l'apparato vestibolare otolitico. La
stimolazione è effettuata in senso orario e antiorario. L'analisi del nistagmo
indotto da questa stimolazione permette di analizzare il funzionamento dei
sistemi otolitici di destra (rotazione oraria) e di sinistra (rotazione
antioraria). La risposta oculare dovuta a questa stimolazione è un nistagmo
complesso che comprende una componente orizzontale, una componente verticale e
una componente di torsione del Fig.15 C bulbo
oculare. Le risposte sono registrate sul piano orizzontale e sul piano
verticale: le velocità delle fasi lente del nistagmo descrivono una modulazione
in funzione del ciclo di rotazione della sedia. I parametri misurati sono
l'ampiezza della modulazione delle componenti verticali e orizzontali e la
componente continua (o bias che corrisponde alla deviazione della media della
sinusoide rispetto allo zero).
La risposta vestibolo-oculare è registrata, al buio,
con elettronistagmografia o video-oculografia. La sedia Fig.
15 C
è sottoposta a un'accelerazione rotatoria breve, quindi a un movimento rotatorio
costante (60°/secondo) in base a un asse verticale; vengono quindi stimolati i
canali semicircolari orizzontali. A velocità di rotazione costante, la risposta
canalare si annulla progressivamente. La sedia è allora inclinata di 13°
rispetto alla gravità, sempre a velocità costante di 60°/secondo ; viene quindi
stimolato, in maniera elettiva, l'apparato vestibolare otolitico. La
stimolazione è effettuata in senso orario e antiorario. L'analisi del nistagmo
indotto da questa stimolazione permette di analizzare il funzionamento dei
sistemi otolitici di destra (rotazione oraria) e di sinistra (rotazione
antioraria). La risposta oculare dovuta a questa stimolazione è un nistagmo
complesso che comprende una componente orizzontale, una componente verticale e
una componente di torsione del Fig.15 C bulbo
oculare. Le risposte sono registrate sul piano orizzontale e sul piano
verticale: le velocità delle fasi lente del nistagmo descrivono una modulazione
in funzione del ciclo di rotazione della sedia. I parametri misurati sono
l'ampiezza della modulazione delle componenti verticali e orizzontali e la
componente continua (o bias che corrisponde alla deviazione della media della
sinusoide rispetto allo zero).
Il test RAIG è utile per definire le patologie del sistema otolitico
In caso di patologie vestibolari acute (neuriti, labirintectomia) poiché i fenomeni di compenso centrale cancellano rapidamente le asimmetrie delle risposte al test RAIG e mascherano in tal modo il deficit otolitico; queste asimmetrie all'esame RAIG permettono di distinguere le malattie vestibolari complete (canalari e otolitiche) o parziali (solo canalari); queste ultime riescono a recuperare più facilmente;
In caso di interessamento vestibolare fluttuante, dove il carattere fluttuante della lesione impedisce il compenso centrale; per esempio, nelle fistole perilinfatiche traumatiche, il Il test RAIG evidenzia una preponderanza direzionale diretta verso il lato leso, segnale di irritabilità otolitica che può indicare la presenza di una fistola
Questo esame permette di valutare le proprietà dinamiche del riflesso otolito-oculare. Darlot C., 1988 ;. Esso consiste nel misurare i movimenti oculari indotti da una rotazione a velocità costante intorno a un asse inclinato rispetto alla gravità. Durante questa stimolazione a velocità costante, i canali semicircolari non sono stimolati. Al contrario, la componente del vettore gravitario (G), che corrisponde alla proiezione di questo sul piano degli organi otolitici, varia in maniera sinusoidale durante la rotazione. Ne deriva una stimolazione alternativa delle macule otolitiche, che provocherà un nistagmo orizzontale complesso. Il test RAIG stimola i due lati del sistema otolitico. Tuttavia, la stimolazione è più importante per il sistema otolitico destro quando la rotazione è in senso orario, e più importante per il sistema otolitico sinistro quando la rotazione è in senso antiorario.
In pratica, il paziente è seduto nell'oscurità su una sedia che ruota a 60°/s secondo un asse inclinato (da 9° a 13° secondo i paradigmi) e i movimenti oculari sono misurati con l'aiuto dell'elettronistagmografia. La velocità della fase lenta orizzontale evocata dalla stimolazione ha due componenti: una distorsione (che corrisponde alla velocità media della
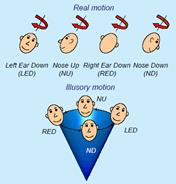
Fig. 16
fase lenta del movimento dell'occhio in senso opposto alla velocità di rotazione della testa) e una modulazione sinusoidale della velocità della fase lenta del movimento dell'occhio, la cui periodicità è identica al periodo della rotazione.
Il test RAIG contribuisce alla diagnosi di una lesione vestibolare monolaterale acuta e le asimmetrie di risposta indicano il lato leso. Così, nelle neuriti vestibolari, e come il test dei potenziali evocati otolitici, esso permette di precisare l'interessamento globale o parziale del nervo vestibolare. Darlot C.,1997.Tuttavia, dopo compenso centrale del deficit, le asimmetrie delle risposte oculari si riducono e questo esame perde il suo interesse.
In caso di interessamento vestibolare bilaterale, permette di valutare la presenza o meno di una funzione otolitica residua. È dunque particolarmente interessante nel bambino in caso di ritardo di acquisizioni motorie, e in particolare di acquisizione della deambulazione, per precisarne l'origine. Wiener-Vacher S.2001.
Gli astronauti hanno riferito di episodi di disorientamento relativamente frequente in condizioni di microgravità e al ritorno in terra. Tali episodi hanno il potenziale di interferire con le prestazioni di un astronauta e di compromettere la sicurezza. Il laboratorio di OVAR principalmente riguarda la comprensione dei meccanismi con cui i giudizi di orientamento sono realizzati al fine di comprendere le cause degli episodi di disorientamento e si spera di fornire il sistema per eliminare tali episodi. Il laboratorio utilizza compiti oculomotori e percettivi per misurare dove gli astronauti si sentono di essere in uno spazio tridimensionale e in quale direzione si sentono muoversi . Questa posizione soggettiva è chiamata egocenter e la posizione e la direzione è determinata sia dagli Otolite e dagli ingressi somatosensoriali.
Il rotatore per il Test della rotazione intorno a un asse inclinato (Off-Vertical Axis Rotator )permette ai ricercatori di studiare i movimenti oculari e la percezione del movimento mentre cambia continuamente l'orientamento del soggetto rispetto alla gravità. Questo dispositivo è attualmente utilizzato per esaminare le modifiche adattive nelle risposte mediata degli Otoliti- seguendo voli Shuttle di breve durata (DSO 499), così come esaminando le interazioni vestibolare autonomiche.
PROVE PENDOLARI AD ACCELERAZIONE DECELLERAZIONE SINUSOIDALE
1e)Test pendolare rotatorio sinusoidale smorzato stimolazione
mantenuta(Grenier)
2e)Test pendolare sinusoidale a scansione di frequenza
3e)Test ad accelerazione sinusoidale
1e) La prova di stimolazione rotoacceleratoria pendolare o sinusoidale o Test pendolare rotatorio sinusoidale smorzato (Grenier)
La prova di stimolazione pendolare è stata applicata per la prima volta da Kreidl nel 1872 per lo studio della reflettività labirintica ed è entrata nella routine otoneurologica qualche decennio fa (1955) per merito di Hennebert e successivamente di Greiner e Conraux (1960). Questa prova si rifà al principio del pendolo di torsione, un sistema fisico che genera delle oscillazioni rotatorie sinusoidali di periodo costante e di ampiezza progressivamente ridotta. La risposta nistagmica riflette il grado di attivazione dei recettori cupolari dei canali semicircolari laterali dell’uno e dell’altro labirinto in rapporto alla dinamica della corrente endolinfatica. Durante l’oscillazione della sedia in senso orario, il flusso dell’endolinfa nel canale semicircolare laterale di destra sarà diretto in senso ampullopeto, durante l’oscillazione in senso antiorario il flusso dell’endolinfa sarà invece ampullipeto in quello di Sinistra.
Per eseguire la stimolazione, il paziente viene sistemato sulla sedia col capo flesso in avanti di 30° e ben fissato per impedirne ogni movimento. Gli occhi rimangono aperti sotto gli occhiali di Bartels o di Frenzel. Durante la rotazione, l’ambiente deve essere completamente oscuro e silente per evitare fenomeni d’interferenza d’origine otticocinetica e audio cinetica. Dopo aver ruotato la sedia dalla sua posizione normale di equilibrio fino a raggiungere un angolo di ampiezza di 180°, la si lascia libera di compiere una serie di oscillazioni sinusoidali dirette alternativamente in senso orario e antiorario. Tali oscillazioni, a causa dello smorzamento impresso alla sedia, tendono nel corso di 15 periodi, ciascuno della durata di 20 sec., a ridursi progressivamente di ampiezza. Sul grafico, all’inizio della oscillazione della sedia, compare una reazione oculomotoria documentata da una deviazione cupoliforme della penna scrivente. Tale deviazione corrisponde allo spostamento tonico degli occhi che si muovono lentamente nella direzione contraria al senso della rotazione. Successivamente, compaiono sul grafico le prime scosse del nistagmo che, dopo 2-3 sec., raggiungono il valore massimo sia di ampiezza che della velocità angolare della fase lenta. Questi valori permangono tali per 2/3 della durata dell’oscillazione della sedia. Poi si riducono in modo rapido e alla fine della oscillazione il nistagmo cessa completamente. Con l’inizio della successiva rotazione della sedia nella direzione opposta, ricompaiono sul grafico il movimento tonico degli occhi, già in precedenza osservato, ma di opposta polarità e successivamente le scosse nistagmiche che si succedono con le stesse caratteristiche di quelle precedenti (Fig. 17).
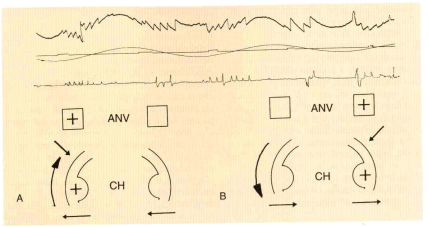
![]() Fig.
17
- Metodo di stim. labirintica rotoacceleratoria pendolare sul piano
orizzontale. A) L’accelerazione ang. positiva da rot. oraria determina una
depolarizzazione del recettore ampollare del can. sem. lat. di destra e
l’attivazione dell’area nucleare omolaterale (ANV) con la comparsa di un
nistagmo diretto verso destra( ) B) Analoga è la reazione vestibolare
a seguito di una stim. rotatoria antioraria ma di segno opposto (
Fig.
17
- Metodo di stim. labirintica rotoacceleratoria pendolare sul piano
orizzontale. A) L’accelerazione ang. positiva da rot. oraria determina una
depolarizzazione del recettore ampollare del can. sem. lat. di destra e
l’attivazione dell’area nucleare omolaterale (ANV) con la comparsa di un
nistagmo diretto verso destra( ) B) Analoga è la reazione vestibolare
a seguito di una stim. rotatoria antioraria ma di segno opposto (![]() ).
(da Greiner G., Conraux C., Collard M., 1969). da Babighian-Otoneurologia-
Piccin- 2008.
).
(da Greiner G., Conraux C., Collard M., 1969). da Babighian-Otoneurologia-
Piccin- 2008.
Esso presenta le seguenti caratteristiche: un periodo di 20 secondi (frequenza 0,05 Hz), un'ampiezza massima di 180°, uno smorzamento esponenziale in 15 periodi, un'accelerazione in partenza di 18°/s2. Il movimento oculare è registrato contemporaneamente al movimento della poltrona. Il parametro studiato è il più delle volte la velocità media della fase lenta del nistagmo, calcolata mediante computer, o l'ampiezza cumulativa del movimento dell'occhio. Quest'ultima è ottenuta addizionando le fasi lente e sopprimendo le fasi rapide. Il tracciato cumulativo ha allora la forma di un sinusoide smorzato sovrapponibile al movimento della poltrona. Sono allora misurati il guadagno e la fase del RVOH. Nel soggetto normale, essi sono rispettivamente vicini a 0,6 per il guadagno e a 10° per la fase. Questo test ha il vantaggio di essere rapido, ma esamina il sistema vestibolare soltanto nel campo delle risposte a basse frequenze, mentre può rispondere a un'ampia gamma di frequenze che vanno da 0,01 Hz a 20 Hz.
Si studia anche nel corso di questo test l'indice di inibizione del nistagmo con fissazione oculare (IFO). In questo caso, si chiede al soggetto di fissare un bersaglio luminoso posto nella maschera di videonistagmografia mentre è sottoposto a questa rotazione. La fissazione oculare induce un'inibizione di oltre il 50% del guadagno del RVOH. Un IFO superiore al 50% è sempre segno di una patologia vestibolare centrale, ma non ha un valore localizzatorio preciso.
In caso di distruzione acuta e unilaterale di un labirinto, si osserva una diminuzione bilaterale del guadagno del RVOH a questo test a basse frequenze, più marcata per le rotazioni verso il lato leso che per quelle verso il lato sano. A distanza, queste alterazioni a basse frequenze (0,05 Hz) scompaiono il più delle volte grazie al compenso vestibolare centrale il che, al contrario delle prove caloriche, limita l'interesse di questo esame quando è eseguito a distanza dalla lesione.
Fig.18
a
velocità angolare
progressivamente
smorzata
Fig 2-29 a -
Elettro nistagmogramma pendolare di un soggetto normale I due estratti della
curva sono stati rilevati verso i 90 di ampiezza massima del quadro all’inizio
della prova (tracciato superiore) e in prossimità della soglia, verso il
termine della prova (tracciato inferiore) La velocità di scorrimento della
carta è di l5mm al secondo Tarawra dei movimenti oculari: lOmm per 10’ di
ampiezza del globo oculare
Su ciascun tracciato, dall’alto in basso:
- la registrazione dei movimenti orizzontali a corrente alternata (A.C )
Costante di tempo: 0,1 Le deflessioni dell’occhio a destra si iscrivono verso
l’alto; le deflessioni a sinistra verso il basso;
- la registrazione dei movimenti orizzontali a corrente continua (CC ) Stessa
convenzione per il senso del nistagmo;
- il movimento della sedia e la linea isoelettrica, posizione di equilibrio del
quadro pendolare;
- la registrazione dei movimenti verticali in corrente alternata (A C 0,1) I
movimenti verticali superiori sono registrati verso l’alto, i movimenti
verticali inferiori verso il basso.
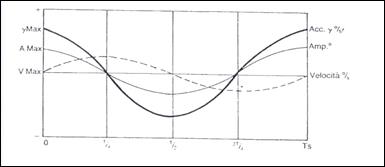
Fig. 19
a velocità angolare
costante
La prova viene condotta oltre che con accelerazioni sinusoidali di valore sopraliminare anche con valori liminari (ampiezza massima dell’oscillazione della sedia di 10°).
Video-Head Impulse Test (vHit): Figura 20 A, B,C
|
|
|
|
|
|
|
|
Impulse Testing DA NOI UTILIZZATO |

Con l'introduzione del video Testa (vHIT), i medici hanno un "amico dei bambini", strumento relativamente facile da usare, e semplice per valutare ciascuno dei 6 canali semicircolari. Durante il test, il bambino indossa un piccolo occhiale leggero, seduti da soli o in braccio a un adulto (Figura 1). Il compito del bambino è semplicemente a fissarsi su un bersaglio. Obiettivi variano con l'età del bambino e possono includere una faccina sorridente adesivo sul muro per un bambino più grande, o un giocattolo attiva che si accende e si muove per un bambino più piccolo.
Come il bambino si fissa sul bersaglio, la loro testa è rapidamente, delicatamente, e saldamente guidata lungo i tre piani di movimento: 1) laterali, 2) a destra anteriore / posteriore sinistra (RALP), e 3) anterior sinistra / posteriore destra (GRV ). Il test richiede solo pochi minuti, la generazione di dati circa il guadagno funzionale di ogni SCC testata, che documenta che gli aerei corretti sono stati testati, e dimostrando la presenza di eventuali saccadi correttive che si sono verificati. Cioè, vHIT determina oggettivamente se una delle sei SCC non funzionano correttamente e permette la registrazione e la misurazione di qualsiasi nistagmo spontaneo o posizionale.
Il vHIT valuta il riflesso oculare vestibolare durante il movimento tipico della testa, ed è sensibile attraverso il normale range di movimento della testa. Durante questa prova, il paziente indossa un video occhiali occhiali-come e mantiene il suo fuoco visivo su un bersaglio fisso, mentre l'esaminatore fa alcuni piccoli ma veloci movimenti della testa. Questo test consente di valutare la funzione vestibolare di un paziente di tutti i canali semicircolari. Usiamo il dispositivo state-of-the-art che non è invasivo e ben tollerato dai bambini che possono seguire le istruzioni di base.
Test della funzione otolitica
Test di rotazione inclinata rispetto alla gravità (Figura 14-5 A, B)
Test OVAR o test RAIG: la poltrona girevole sulla quale il soggetto è seduto nell'oscurità è ruotata a velocità costante secondo un asse inclinato rispetto alla gravità. Questo movimento produce una stimolazione generale del sistema otolitico e in ritorno una risposta vestibolo-oculare che è registrata e misurata. [Wiener-Vacher et al 1994/6/2001]
Misurazione della verticale soggettiva (Figura 15)
 Questa prova è usata per
valutare la funzione del otricolo, un altro sensore otolite nell'orecchio
interno È una misura che valuta la precisione con cui un soggetto seduto
nell'oscurità può porre in posizione verticale una barra fosforescente con
l'aiuto di un comando a distanza con una precisione di +/- 2°. Questo test è
applicabile solo ai bambini di età superiore a 7-8 anni. I bambini più piccoli
non hanno la nozione di verticale fino all'età di 2-3 anni e non possono quindi
eseguire il compito, i bambini di 4-8 anni sono molto imprecisi nel riprodurre
la verticale in tali condizioni (precisione di +/- 4°). Una lesione recente
(meno di 3 mesi) della parte otolitica del vestibolo si manifesta con una
deviazione della percezione della verticale soggettiva dal lato della lesione
vestibolare. I fenomeni di compensazione normalizzano in seguito questi
risultati.
Questa prova è usata per
valutare la funzione del otricolo, un altro sensore otolite nell'orecchio
interno È una misura che valuta la precisione con cui un soggetto seduto
nell'oscurità può porre in posizione verticale una barra fosforescente con
l'aiuto di un comando a distanza con una precisione di +/- 2°. Questo test è
applicabile solo ai bambini di età superiore a 7-8 anni. I bambini più piccoli
non hanno la nozione di verticale fino all'età di 2-3 anni e non possono quindi
eseguire il compito, i bambini di 4-8 anni sono molto imprecisi nel riprodurre
la verticale in tali condizioni (precisione di +/- 4°). Una lesione recente
(meno di 3 mesi) della parte otolitica del vestibolo si manifesta con una
deviazione della percezione della verticale soggettiva dal lato della lesione
vestibolare. I fenomeni di compensazione normalizzano in seguito questi
risultati.

Figura 16. Verticale e orizzontale soggettive. Prima dei 4 anni la nozione di verticalità non è compresa, mentre il corpo può riprodurre la verticale. Da 4 a 8 anni la verticalità può essere riprodotta ma con una grande variabilità di valutazione: ± 4°. Dopo gli 8 anni, il test è effettuato con la stessa precisione che nell’adulto: ± 2 °.
Potenziali Vestibolari Evocato Miogenici Cervicale (cVEMPs) o Potenziali Evocati Miogenica oculare (oVEMPs) miogeni sacculo-collici (o PEOM: potenziali evocati otolitici miogeni) (Figura 17 A, B)
 Le
vie vestibolospinali sono stimolate con l'aiuto di impulsi sonori (tone
burst) o di click di forte intensità (da 100 a 110 dB) distribuiti al
livello di ogni orecchio. Si registra l'effetto di questa stimolazione sulla
contrazione attiva dei muscoli sternocleidomastoidei registrata mediante
elettromiografia. Un'assenza di risposta significa una lesione della via sacculospinali.
Si può eseguire questo esame nel bambino non appena egli è capace di mantenere
eretto il capo (a partire dai 3-4 mesi). La contrazione attiva dei muscoli è
ottenuta inclinando all'indietro il bambino che è seduto sulle ginocchia di un
genitore o se è più grande e cooperante (> 5-6 anni) chiedendogli di
appoggiarsi sulla mano dell'esaminatore con il mento. [7] Per
quanto riguarda la valutazione del utricolo e il ramo superiore del nervo
vestibolare, oculare Evocati Miogenica Potenziali (oVEMPs) sono state
registrate nei bambini di 1 mese di età.
Le
vie vestibolospinali sono stimolate con l'aiuto di impulsi sonori (tone
burst) o di click di forte intensità (da 100 a 110 dB) distribuiti al
livello di ogni orecchio. Si registra l'effetto di questa stimolazione sulla
contrazione attiva dei muscoli sternocleidomastoidei registrata mediante
elettromiografia. Un'assenza di risposta significa una lesione della via sacculospinali.
Si può eseguire questo esame nel bambino non appena egli è capace di mantenere
eretto il capo (a partire dai 3-4 mesi). La contrazione attiva dei muscoli è
ottenuta inclinando all'indietro il bambino che è seduto sulle ginocchia di un
genitore o se è più grande e cooperante (> 5-6 anni) chiedendogli di
appoggiarsi sulla mano dell'esaminatore con il mento. [7] Per
quanto riguarda la valutazione del utricolo e il ramo superiore del nervo
vestibolare, oculare Evocati Miogenica Potenziali (oVEMPs) sono state
registrate nei bambini di 1 mese di età.

Test Posturografico ("piattaforma postura")
Durante questa prova, un paziente sta in piedi su una piattaforma e misuriamo come stabile è in condizioni diverse (ad esempio, con gli occhi aperti o chiusi) e in risposta a piccoli movimenti della piattaforma. Questo test consente di valutare la causa di squilibrio
Figura 18
La batteria di test motilità oculare può essere facilmente adattato per pediatria. E 'stato riportato che la stimolazione a tutto campo è necessario invocare una risposta optocinetica involontario (OPK) Dal momento che la risposta OPK è presente dalla nascita, la stimolazione a tutto campo elimina gli eventuali problemi che potrebbero derivare da mancanza di stimoli campo periferico. [Schor C, Narayan V.,1981 ] Incorporando stimoli a tutto campo divertenti nella prova optocinetico può essere particolarmente utile per tenere i bambini interessati a, e partecipare a , l'attività (Figura 19).
|
|
|
Figura 19. Pediatric stimoli optokinetic disponibili in Interacoustics VO425 FireWire® VNG |


È altrettanto importante mantenere l'attenzione del bambino durante il resto della valutazione motilità oculare. Infatti, Cyr.980 suggerito di usare personaggi dei cartoni animati per la calibrazione, nonché per lo svolgimento regolare e test saccade. Chiaramente, se i professionisti mantenere interessante, divertente, e la sperimentazione a base di gioco per i bambini, poi le prove stesse sono meno intimidatorio per il bambino, il bambino è più disposti a impegnarsi in più attività di test, e una valutazione diagnostica più significativa e approfondita può essere completato
Equivalenti emicranici
Bambina di 7 anni.
Da 1 mese, diversi episodi di vertigini , nausea e mal di testa da 1/2 ora a 1 ora, fotofobia.
A scuola, a fine giornata, dopo la televisione, la lettura.
Esame oto-neuro-vestibolare: normale al di fuori di un'ipofunzione canalare destra isolata al test calorico (?).
Scanner: normale.
Esame oftalmologico: ipermetropia e disturbo della convergenza; occhiali e rieducazione ortottica.
Terapia medica antiemicranica: antiinfiammatori non steroidei e antalgici.
Quali sono le cause di vertigine nel bambino?
I DISTURBI VESTIBOLARI SPECIFICI CHE POSSONO VERIFICARSI DURANTE L'INFANZIA COMPRENDONO
Equivalenti emicranici
Tra le diagnosi da ipotizzare in presenza di vertigini del bambino, la più frequente è quella di equivalenti emicranici (25%) (Figura 20 ):
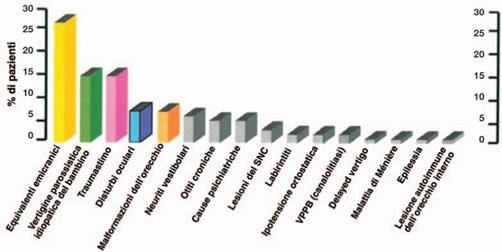 l
l
Figura 20. Diagnosi da ipotizzare in presenza di vertigini del bambino. SNC: sistema nervoso centrale; VPPB: vertigine posizionale parossistica benigna.
la vertigine si associa a cefalee, le precede, le accompagna o insorge in alternanza con le cefalee. Queste vertigini equivalenti emicranici sono spesso mal tollerati (nausea, vomito non sono eccezionali) e possono associarsi a fotofobia (sintomo caratteristico della natura emicranica dell'episodio). Gli episodi di vertiginii equivalenti emicranici possono durare diverse ore, verificandosi più spesso per fatica, alla fine della giornata. Gli esami otologici, vestibolare e neurologico sono rigorosamente normali. La frequenza e l'intensità degli equivalenti emicranici sono spesso aggravate dalla presenza di disturbi oftalmologici (disturbi della rifrazione misconosciuti e non corretti, disturbi della convergenza oculare). [9] Ogni equivalente emicranico dovrebbe subire una valutazione oculistica completa per eliminare questo fattore aggravante. Il trattamento degli equivalenti emicranici del bambino deve prima di tutto iniziare con l'eliminazione dei fattori aggravanti (problemi oftalmologici, igiene di vita, sonno corretto, riduzione dello stress, gestione psicologica se necessario) associata a trattamenti analgesici semplici (paracetamolo, antinfiammatori isolati o in associazione, acido acetilsalicilico). Questo solo atteggiamento terapeutico è spesso sufficiente a ridurre la frequenza e l'intensità delle crisi. Se queste misure preventive sono insufficienti, può essere efficace una terapia con derivati della segale cornuta a basse dosi, che però non può essere somministrata a lungo termine nel bambino. I derivati antiserotoninergici non sono autorizzati prima dell'età di 12 anni e saranno presi in considerazione esclusivamente nei casi refrattari.
L'inizio molto precoce degli episodi (durante i due primi anni di vita), il loro carattere recidivante e regolare (ogni mese od ogni 2 mesi) senza fattore scatenante e molto mal tollerato (vomito) quando esistono dei precedenti familiari di emicrania vera (o di emicranie accompagnate) devono far sospettare un'atassia episodica di tipo II, malattia genetica il cui trattamento è diverso dall'emicrania vera. In effetti in questo caso gli attacchi si possono risolvere con un trattamento con acetazolamide, ma la terapia di lunga durata favorisce in alcuni pazienti la litiasi renale. La risposta a questo trattamento ha attualmente valore diagnostico. Nel bambino si può ricorrere in prima intenzione al trattamento con la l-leucina, molto meglio tollerato e che sembra essere efficace nei bambini piccoli che presentano questa patologia senza avere gli effetti secondari e le complicanze dell'acetazolamide (terapia in corso di valutazione).
La vertigine emicranica equivalente (VEE)
La VE può essere definita come “equivalente” solo quando l’attacco vertiginoso-posturale sostituisce completamente l’attacco di cefalea, presentandosi in forma isolata, senza alcun rapporto temporale diretto con le crisi di cefalea. La vertigine rappresenta un sintomo alternativo alla crisi cefalalgica, scatenato probabilmente da meccanismi patogenetici analoghi e struttura bersaglio diversa.
Nella VEE non esiste uno stretto rapporto temporale tra la singola crisi di cefalea e di vertigine, ma la seconda si verifica quasi sempre nella fase florida come manifestazione sostitutiva, quindi equivalente, della prima. La VEE può essere di vario tipo, in base al rapporto temporale esistente tra il periodo d’esordio della vertigine ed il periodo fondo delle crisi algiche; nella vita del soggetto emicranico la VEE può infatti precedere (VEE precoce), sostituire parzialmente (VEE intercnitica) o seguire (VEE tardiva), il periodo fondo della cefalea emicranica.
La VEE precoce
Si tratta di turbe vertiginoso-posturali che rappresentano un sintomo alternativo (equivalente) alla cefalea, precedendo nel tempo (precoce) il periodo delle crisi algiche (precritica), configurandosi spesso come la prima vera manifestazione clinica dell’habitus emicranico del paziente.
Le due principali forme cliniche di VEE precoce sono rappresentate dal torcicollo parossistico e dalla vertigine parossistica dell’infanzia.
Queste due entità nosologiche sono troppo spesso definite come precursori emicranìci, ossia come prime manifestazioni di un habitus che poi si esplicherà sotto forma di cefalea emicranica, A nostro giudizio, invece, sia la vertigine parossistica che il torcicollo parossistico dell’infanzia hanno già tutte le caratteristiche di una VEE precoce, con dignità clinica e caratteristiche nosologiche proprie: sono pertanto una vera patologia vestibolare emicranica e non un semplice precursore.
La vertigine parossistica infantile si manifesta in modo subacuto e senza prodromi, con malessere ed arresto motorio volontario del bambino che può rimanere sofferente da alcuni minuti a poche ore per poi riprendere normalmente la propria attività motoria ed anche ludica. Il bambino, in età prescolare, non è per lo più in grado di descrivere la propria sintomatologia e spesso queste manifestazioni, proprio per l’arresto motorio, vengono scambiate per assenze epilettiche. Quando il bambino è in grado di descrivere i sintomi, riferisce una vertigine oggettiva rotatoria che è responsabile, insieme alla paura, dell’arresto motorio, Sono quasi sempre presenti il pallore e la sudorazione, mentre
la nausea ed il vomito possono anche mancare. Non abbiamo mai avuto l’occasione di osservare direttamente una fase acuta, ma alcuni genitori hanno riferito di aver notato i movimenti oculari del nistagmo (un padre è riuscito anche a fornire un filmato del nistagmo, che aveva ca— ratteristiche periferiche). La presentazione di questa VEE avviene di solito fra i 3 e gli 8 anni, ma abbiamo avuto anche pazienti con esordio tardivo, comunque sempre prima del periodo cefalalgico. La diagnosi è sempre anamnestica: l’esordio in pieno benessere senza rapporti causali, la durata limitata (da minuti ad un massimo di 2 ore), la risoluzione completa con ripresa della normale attività motoria, la tendenza a recidivare senza alcuna regola temporale (talora una presentazione periodica a grappolo), la negatività audio-vestibolare al di fuori delle crisi e la accertata familiarità emicranica, sono i cardini anamnestici per la diagnosi. La risonanza magnetica è indicata in caso di possibile persistenza post-critica di segni nistagmici o per placare i timori di genito ri ansiosi. La terapia è prevalentemente psicologica: far ben comprendere ai genitori la natura dei disturbi, spiegare loro che i sintomi scompariranno spontaneamente nel tempo, invitarli ad essere rassicuranti verso il bambino al momento della crisi. L’uso dei farmaci per la profilassi emicranica è sconsigliata, vista l’età dei pazienti. E opportuno avvisare i genitori della possibilità che il figlio sviluppi una cefalea emicranica dopo la pubertà.
Il torcicollo parossistico infantile è caratterizzato da episodi di tonica rotazione del capo con flessione (tilt) verso la spalla, della durata da minuti a poche ore, con risoluzione senza esiti organici. Rispetto alla vertigine parossistica infantile, è più raro, si sviluppa più precocemente, tende ad essere più prolungato e gli episodi sono meno numerosi. Nella nostra casistica abbiamo avuto solo due bambini che hanno presentato sia la vertigine che il torcicollo parossistico: in entrambi i casi, il torcicollo era esordito prima della vertigine e si era esaurito con la comparsa della vertigine parossistica. Verosimilmente il torcicollo è espressione di sofferenza emicranica del sistema discendente maculospinale e la vertigine di una analoga sofferenza del sistema ascendente canalare-oculomotorio.
VERTIGINE PAROSSISTICA BENIGNA DEL BAMBINO (20%)
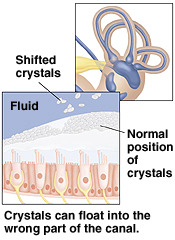 È
la seconda diagnosi da prendere in considerazione in ordine di frequenza. Deve
essere sospettata in particolare nei bambini di 2-3 anni, se e solo se la vertigine
è breve (meno di 10 min) non associata a una cefalea (bambino non
dolente), in genere ben sopportata (a volte pallore, nausea ma raramente
vomito) e priva di ripercussioni sulle attività prima e dopo il periodo di malessere.
. Si tratta di un disturbo comune che accade a circa il 2,6% dei bambini alcuni
esperti ritengono che sia un precursore di emicrania . (Abu-Arufeh e Russell,
1995). A
volte è anche osservato come complicazione chirurgica di impianto cocleare. 1,26,27
È
la seconda diagnosi da prendere in considerazione in ordine di frequenza. Deve
essere sospettata in particolare nei bambini di 2-3 anni, se e solo se la vertigine
è breve (meno di 10 min) non associata a una cefalea (bambino non
dolente), in genere ben sopportata (a volte pallore, nausea ma raramente
vomito) e priva di ripercussioni sulle attività prima e dopo il periodo di malessere.
. Si tratta di un disturbo comune che accade a circa il 2,6% dei bambini alcuni
esperti ritengono che sia un precursore di emicrania . (Abu-Arufeh e Russell,
1995). A
volte è anche osservato come complicazione chirurgica di impianto cocleare. 1,26,27
DIAGNOSI
Per una diagnosi di vertigine parossistica benigna dell'infanzia da apportare al bambino deve avere:
•almeno cinque attacchi.
•ottenere vertigini (capogiri) un sacco, che succede tutto ad un tratto, e andare via dopo pochi minuti ad alcune ore.
•un normale esame neurologico: test per assicurarsi che non vi è alcun problema nel cervello
•funzioni audiometrici e vestibolari normale tra gli attacchi: non c'è problema che può essere trovato nell'orecchio interno (parte interna delle orecchie).
•normale elettroencefalografia (EEG): il cervello di una persona fa piccole quantità di energia elettrica e di questo test fa in modo che questo sta lavorando bene.
Adattato da:. L'Headache Classification sottocommissione della International Headache Society 2004 [3]
La diagnosi differenziale
Le diagnosi differenziali sono diversi disturbi medici che possono causare gli stessi sintomi. Prima che un medico fa una diagnosi finale , che significa che sono sicuri di ciò che disturbo medico sta causando il problema, pensano di ciò che altre condizioni mediche hanno quasi gli stessi sintomi stessi o, e assicurarsi che non è uno di loro. [4]
Alcune delle diagnosi differenziali di vertigine parossistica benigna dell'infanzia sono:
•fossa posteriore tumori: questo un tumore nella parte del cervello in prossimità del fondo del cranio. Tumori della fossa posteriore accadono più nei bambini che fanno in adulti. Circa il 54% -70% di tutti i tumori cerebrali che i bambini iniziano nella fossa posteriore.
•anomalie della colonna vertebrale cervicale: questi sono problemi con la parte della colonna vertebrale nel collo.
•Patologie otologiche: questi sono problemi nella parte interna di orecchie.
•epilessia (l'epilessia occipitale benigna): questo è un tipo di malattia del cervello.
•Disturbi metabolici: questo è quando c'è un problema con il modo in cui il corpo produce energia dal cibo.
L'esame otologico, vestibolare e neurologico clinico è normale. Gli episodi si possono ripetere per qualche mese, o 1 anno, e scompaiono spontaneamente. Se questi sintomi insorgono in bambini un poco più grandi (4-5 anni o più), è fondamentale ricercare un'altra causa e in particolare un disturbo oftalmologico prima di considerare questa diagnosi. In genere non è necessario alcun trattamento. È importante informare i genitori della benignità di questa affezione di origine sconosciuta e chiedere loro di ritornare se i sintomi non si risolvono o si modificano. Non è utile eseguire una RM in questi casi particolari dove gli esami otologici neurologici e vestibolari sono normali; la RM non è priva di rischi a questa età (è eseguita in anestesia generale ed è relativamente traumatizzante) ed è costosa, mentre è sempre normale in questi casi. Vertigine parossistica benigna dell'infanzia (BPVC in breve) (questo significa vertigini innocuo, che avviene ancora e ancora e accade all'improvviso) è una condizione medica che si verifica di solito nei bambini a partire dai due ai cinque anni di età; scompare spesso dall'età di otto anni. Ma BPVC può iniziare giovane come un paio di mesi al più tardi a 12 anni di età. Essa provoca una sorta di vertigine chiamata vertigine parossistica . Con questo tipo di vertigine persone sentono che sono in movimento o rotazione o che l'interno della loro testa è in movimento o spinning.
Vertigine parossistica benigna
Maschio di 2 anni e 1/2.
Quattro episodi ricorrenti di vertigini: dice «la casa gira».
Durata breve: alcuni secondi, meno di 10 minuti.
Si aggrappa o si siede.
Movimenti oculari (nistagmo?) talora osservati.
Molto ben tollerato: non dolori, non nausee, non vomiti?
Gioca normalmente in seguito.
Esame oto-neuro-vestibolare: normale.
Richiesta di esame oftalmologico, non di diagnostica per immagini, ma rivedere al termine di 4 mesi o prima se vi fosse un cambiamento dei segni.
Rivisto al 4°mese: esame normale.
Scomparsa spontanea delle vertigini in 6 mesi, nessun trattamento.
Quadro di trauma cranico (10%)
Le vertigini devono essere prese molto sul serio perché possono essere il segno di una frattura della rocca petrosa e/o di una fistola perilinfatica. [10] Una frattura o fessura del guscio dell'orecchio interno, permettendo la fuoriuscita di liquidi dell'orecchio interno, può portare a sordità evolutiva e areflessia vestibolare se non è effettuato rapidamente un intervento chirurgico di riempimento di questa breccia. Una tale fistola espone il bambino a meningiti recidivanti. Ogni bambino che ha una turba dell'equilibrio (atassia, deviazione posturale) o una vertigine e in più, presenta una otorragia e una ipoacusia nell'immediato periodo successivo a un trauma cranico dovrebbe almeno essere sottoposto a un esame otorinolaringoiatrico in urgenza (audiometria ed esame otovestibolare) per verificare la presenza di segni di frattura della rocca (emottisi, otorragia, sordità di trasmissione, di percezione o mista, areflessia o nistagmo da lesione o da irritazione vestibolare). Questi segni dovranno portare alla richiesta di una TC in urgenza alla ricerca:
• di una linea di frattura sulla squama temporale che attraversa o meno l'orecchio interno, non sempre evidente nel bambino;
• di un'immagine aerea nel labirinto, che indica l'apertura dell'orecchio interno nell'orecchio medio (segno molto transitorio, che scompare in meno di 8 giorni) e la presenza di una fistola che deve essere esplorata chirurgicamente in urgenza.
È interessante di sapere che nel bambino la rima di frattura può non essere visibile e tuttavia essere associata a un interessamento funzionale vestibolare o uditivo importante per emorragia intralabirintica o contusione labirintica.
Una lesione funzionale uditiva e/o vestibolare che non dimostra una fistola o un trauma della catena ossiculare dovrà essere rivista entro 8-10 giorni per un nuovo esame audiometrico e vestibolare al fine di valutare l'evolutività delle lesioni; l'aggravamento dei deficit audiovestibolari porta a sospettare un processo evolutivo e una fistola e porta a un'esplorazione chirurgica, mentre la stabilità dei deficit o il loro miglioramento fanno porre la diagnosi di sequela o di contusione semplice. La stabilità, o il miglioramento dei deficit, sarà il segno di una contusione dell'orecchio interno in fase di recupero. Il bambino deve essere oggetto di controlli regolari per confermare questa evoluzione favorevole.
rilevare la fistola perilinfatica, evitare il peggioramento uditivo, limitare il rischio di meningite
Bambina di 6 anni.
Caduta da un mezzanino, perdita di coscienza + trauma cranico + otorragia destra, emotimpano destro, vertigini per 48 ore, urgenza: TC normale, non controllo.
10 giorni dopo: torcicollo, ipoacusia, non vertigini.
Esame: emotimpano, assenza di riflesso dello stapedio destro, sordità mista destra, areflessia vestibolare destra al test calorico, non compensata, segno di Tullio destro ai suoni forti (120 dB, 1kHz), preponderanza direzionale destra nel test RAIG: segno di irritabilità otolitica.
revisione della prima TC: pneumolabirinto, sparito alla seconda TC.
Esplorazione chirurgica: fessura palatina, riempimento senza stapedectomia.
Evoluzione: recupero completo uditivo e vestibolare, stabile.
La malattia di Ménière è meno comune nei bambini. Si tratta di un disturbo periferico coinvolge uno squilibrio di un fluido dell'orecchio interno chiamato endolymph. I sintomi includono perdita fluttuante dell'udito, tinnito, pressione auricolare o sensazioni di pienezza, e gli episodi di vertigine e nausea.
Alcuni disturbi vestibolari meno comuni nei bambini includono questi disturbi periferici: fistola perilinfatica (una lacrima nelle membrane delle finestre ovali o rotonde dell'orecchio interno), e la sindrome dell'acquedotto vestibolare allargato (un anormalmente grande tubo di collegamento endolymph dell'orecchio interno al sacco endolinfatico)
Disturbi visivi
Disturbi oftalmologici
Segni: cadute frequenti (meno di 5 anni), vertigini i dopo i tentativi di lettura, videogiochi e televisione, a scuola, in fine di giornata (oltre i 5 anni).
Mai una grande vertigine permanente.
Spesso cefalee (associate o in alternanza), a volte solo sensazioni vertiginose; spesso terreno emicranico.
Esame oto-neuro-vestibolare normale.
Talvolta, tuttavia, visualizzazione di un disturbo di convergenza.
Non diagnostica per immagini.
Richiesta di bilancio oftalmologico: misurazione della rifrazione sotto dilatatori (Ciclolux®) e bilancio ortottico.
La terapia è oftalmologica: uso degli occhiali e/o rieducazione ortottica.
Nel 10% dei casi, le vertigini del bambino a partire dall'età di 5-6 anni non hanno come sola e unica causa dei disturbi visivi. [Anoh-Tanon et al.,2000; Bucci 2003/4] La correzione di questi disturbi visivi (uso degli occhiali e/o rieducazione ortottica) da sola fa scomparire i disturbi. I disturbi visivi possono essere dei disturbi di rifrazione (miopia, ipermetropia, astigmatismo) o delle anomalie della vergenza oculare. Le sensazioni di vertigine sono delle sensazioni di rotazione o di oscillazione, brevi ma ripetute, spesso legate alla stanchezza (a scuola, alla fine della giornata, dopo sedute prolungate al computer o alla televisione, sedute prolungate di lettura), possono insorgere a volte all'addormentamento o al risveglio il mattino. Queste sensazioni possono essere isolate o associate a cefalee, a nausea ma raramente a vomito. Insorgono spesso in bambini che hanno un terreno emicranico. L'esame può evidenziare un difetto di convergenza oculare facendo fissare un piccolo bersaglio (disegno su un abbassalingua per esempio) mentre lo si avvicina progressivamente sulla linea mediana della radice del naso. L'espressione clinica di questi problemi con vertiginii è attualmente in aumento nei bambini dato lo sviluppo delle attività ricreative che utilizzano degli schermi televisivi o dei computer, dei videogiochi che richiedono degli sforzi di fusione oculare importanti e prolungati spesso per molte ore. In questi casi solamente il controllo oftalmologico (valutazione della rifrazione sotto dilatatori per eliminare l'effetto correttivo dei processi accomodativi, molto efficaci nel bambino piccolo, e bilancio ortottico eseguito da un ortottista) è anormale. Non saranno la RM o la TC ad aiutare nella diagnosi e portare al solo trattamento adatto, che è l'uso di occhiali e/o la rieducazione ortottica.
Neurite vestibolare
NEURITE VESTIBOLARE
infiammazione acuta del settore vestibolare non associato a manifestazioni a carico del labirinto cocleare nè a carico del sistema nervoso centrale
SINTOMATOLOGIA
Vertigine ad esordio improvviso (nel 73%) dei casi
Nistagmo nel 68% dei casi (orizzontale o rotatorio)
Assenza di sintomi “otologici” cocleari (NO ipoacusia, acufeni): FUNZ UDITIVA NORMALE
Assenza di sintomi e segni neurologici associati
Preceduto (o in concomitanza) da evento infettivo quale: flogosi alte vie aeree, tonsillite, sinusite, sepsi dentale per cui sospetta origine virale (ESATTA EZIOLOGIA: SCONOSCIUTA!)
Spesso si verificano piccole epidemie: EPIDEMIC VESTIBULAR NEURONITIS (prima in Svizzera a Ginevra nel 1888 !! “The Staggers” di Chelsea nel 1949)
EPIDEMIC VERTIGO IN CHILDREN Preceduto (o in concomitanza) da evento infettivo quale: flogosi alte vie aeree, tonsillite, sinusite, sepsi dentale per cui sospetta origine virale (ESATTA EZIOLOGIA: SCONOSCIUTA!)
Spesso si verificano piccole epidemie: EPIDEMIC VESTIBULAR NEURONITIS (prima in Svizzera a Ginevra nel 1888 !! “The Staggers” di Chelsea nel 1949)
di dimostrare l'assenza di interessamento dell'udito e di quantificare il danno vestibolare. Se questo è rilevante, le possibilità di recupero sono minime, se la lesione è parziale si osserva il 25% di recupero con un ritorno a una funzione vestibolare Una grande vertigine rotatoria con vomito nel decorso di un episodio infettivo virale deve far pensare a una neurite vestibolare (5% delle vertigini del bambino). Il quadro può essere ingannevole nei bambini piccoli che non si esprimono ancora chiaramente e dove la vertigine si manifesta solo con vomito, dolori addominali, quadro che può mimare completamente quello di una gastroenterite. Tuttavia all'esame clinico esistono dei segni associati che non ingannano se vengono ricercati: la perdita di equilibrio sempre dallo stesso lato quando il bambino si tiene in piedi sul materasso o sul suolo, a occhi chiusi, la presenza di nistagmo (a condizione che sia ricercato senza la fissazione dello sguardo, con occhiali di Frenzel). Tali constatazioni devono far prescrivere una valutazione audiovestibolare. Questa valutazione permette normale o subnormale in poche settimane o mesi. In ogni modo si ottiene una compensazione, tanto più rapida quanto più il bambino è mobilizzato rapidamente e senza trattamento vestiboloplegico. Una RM potrebbe mostrare un ipersegnale a livello del nervo vestibolare, ma questo segno è molto incostante. Il trattamento di queste neuriti è soprattutto sintomatico, per limitare nausea e vomito e facilitare il compenso centrale del deficit vestibolare, attraverso la rieducazione vestibolare, che nel bambino deve essere attuata sotto forma di giochi (giochi con la palla, raccolta di giocattoli al suolo, movimenti rapidi della testa con fissazione visiva di riferimenti fissi).
La Neurite vestibolare provoca vertigini e labirintite causa sia vertigini e sintomi acustici. Entrambi derivare da infezioni dell'orecchio gravi e possono causare sintomi acuti di vertigine e nausea che di solito, ma non sempre, regrediscono entro 4-6 settimane. Questi disturbi sono attribuiti ad una infezione virale del ganglio trigeminale, un insieme di cellule nervose situate proprio dietro l'orecchio, (neurite vestibolare) o infezione batterica o virale dell'orecchio interno (labirintite). –
Labirintite
La diagnosi di neurite sarà differenziata da quella di labirintite con l'esame otoscopico che riscontra, in caso di labirintite, un'otite media acuta purulenta. In questo caso, il trattamento comporta una paracentesi per un prelievo batteriologico, una terapia antibiotica a largo spettro per via endovenosa condotta in ambiente ospedaliero per evitare la distruzione cocleovestibolare e l'estensione dell'infezione (mastoidite, paralisi faciale, setticemia, meningite e localizzazioni settiche extrapetrose). L'udito e la funzione vestibolare sono purtroppo compromessi gravemente e spesso è inevitabile una cofosi con areflessia vestibolare nonostante una terapia precoce.
EPIDEMIC VERTIGO IN CHILDREN Williams S. Epidemic vertigo in children. Med J Aust 1963;2; 6-12
C W Cooper, Vestibular neuronitis: a review.., British Journal of General Practice, 1998,43,164-167
Zannoli R., A child with vestibular neuritis. Is Adenovirus implicated? Brain & Development 28 (2006) 410-412
Compensazione dei deficit vestibolari nel bambino
Il bambino tollera e compensa molto rapidamente un deficit vestibolare acuto monolaterale.
Attenzione alle compensazioni ritardate, segno di evolutività della lesione dell'orecchio interno.
Bambino di 3 anni, angina febbrile, dolori addominali, vomito: diagnosi di gastroenterite nel reparto d'urgenza.
15 o giorno, lamenta ipoacusia sinistra (al telefono): cofosi sinistra; TC normale; diagnosi di neurite.
Quarto mese, visto a Parigi dopo trasloco; visita di controllo: deficit vestibolare non compensato... Al 13° mese, idem; nuova TC: labirintite ossificante.
Tumore della fossa posteriore
La diagnosi di tumore della fossa posteriore, tanto temuta in presenza di vertigini del bambino, è rara: meno dell'1%. I tumori della fossa posteriore sono piuttosto responsabili di disturbi dell'equilibrio che non di vertigini , e soprattutto sono sempre associati ad altri segni neurologici. È per questo motivo che l'esame vestibolare clinico di un bambino portatore di vertigini o di disturbi dell'equilibrio deve sempre essere completato da un esame dell'oculomotricità e da un esame neurologico (ricerca di una sindrome cerebellare, valutazione di nervi cranici, motilità, sensibilità profonda, riflessi osteotendinei, segni piramidali). Il minimo segno neurologico di localizzazione (lesione audiovestibolare, torcicollo permanente, paralisi faciale oculomotoria, emianopsia, segno di Babinski, ipertono monolaterale…) deve portare alla richiesta di una TC e di una RM. Nel bambino i tumori più frequenti sono quelli della fossa posteriore con gli astrocitomi. La loro diagnosi precoce permette un'exeresi chirurgica che può essere completa allo stadio precoce e ottenere la guarigione.
La patologia neurologica di interesse vestibolare
PSEUDOTUMOR CEREBRI CASANI
L il termine usato per la prima volta da onne (1914) per riferire una ipertensione endocranica senza tumore, Varie altre denominazioni sono state successivamente proposte per indicare la suddetta condizione e tra queste quelle che hanno avuto maggior diffusione sono “Ipertensione endocranica benigna” ed ultimamente “Ipertensione intracranica idiopatica” dizione quest’ultima non scevra di critiche, ma ufficialmente adottata e costantemente utilizzata dalla comunità scientifica anglosassone con la sigla IIH (Idiopathic Intracranial Hypertension). Con questi termini attualmente si definisce una sindrome da ipertensione intracranica manometricamente accertata e con decorso benigno (salvo un rischio di cecità nel 10-30% dei pazienti), decorrente senza alterazioni liquorali né sintomi neurologici localizzatori (tranne papilledema ed occasionale paresi del VI nervo cranico) o disturbi della coscienza, e con neuroimmagini negative per presenza di tumori, patologia venosa o dilatazione ventricolare. La diagnosi di questa sindrome quindi può essere posta solo dopo aver escluso qualsiasi altra causa di ipertensione endocranica ed inoltre dopo aver eseguito una rachicentesi per documentare la normalità dei parametri liquorali. La eziologia è ignota pur essendo accertato che questa malattia può essere determinata da alcuni farmaci (contraccettivi orali, eccesso di vitamina A, tetracicline, minociclina, ormone della crescita e vari altri) e verificarsi in corso di affezioni sistemiche infiammatorie, dismetabolismi ed in particolare nell’obesità. L’incidenza annuale è stimata di 1-2 casi per 100.000 abitanti ed aumenta a 20 per 100.000 nel gruppo ad alto rischio di giovani donne obese, La sintomatologia si instaura generalmente nell’arco di alcune settimane ma esistono anche forme acute e croniche.
Le cefalee sono il sintomo più frequente e, nel corso della malattia, possono interessare sino al 99% dei pazienti ed altrettanto frequente (98-100%) è l’edema della papilla ottica, solitamente ma non sempre bilaterale e generalmente caratterizzato da una lenta evoluzione sino all’atrofia ottica. Questa alterazione spesso non è accusata dai pazienti salvo che per la comparsa brusca di transitorie eclissi visit’e con completa cecità. La diplopia si riscontra in circa un terzo dei casi e per lo più è riferibile a paralisi uni o bilaterale dell’abducente. Molto frequenti sono anche i sintomi di interesse audiovestibolare come gli episodi di disequilibrio, i deficit uditivi e gli acufeni, disturbi che essendo spesso associati nello stesso soggetto e non raramente presenti all’esordio della malattia fanno si che non raramente il primo approccio medico con questi pazienti avvenga proprio da parte dell’otolaringologo (Murphy, 1991).
Gli acufeni sono il più spesso intermittenti ed unilaterali e di solito con carattere pulsante (Sismanis et al., 1985). Resta infine da ricordare che esistono in letteratura alcuni studi (Rudnick e Sismanis, 2005) che descrivono la presenza di rinoliquorrea in soggetti affetti da ipertensione endocranica idiopatica e questo dato appare decisamente impegnativo a focalizzare l’inchiesta anamnestica anche su domande che permettano di escludere la coesistenza di questa potenzialmente grave patologia.
Restano (la considerare alcune malattie nelle quali la Compressione/ischemia sui centri vestibolari troncoencefalici si verifica attraverso alterazioni della dinamica del liquido cerebrospinale che inducono una dilatazione ventricolare attiva (idrocefalo) con conseguente sofferenza del tessuto nervoso circostante. L’aumento del volume dei ventricoli, oltre che per meccanismo ex-vacuo per atrofia del parenchima cerebrale circostante, può determinarsi per ostacoli alla sua circolazione a vari livelli (ivi comprese le strutture deputate al suo riassorbimento) dovuti a cause assai diversificate, per lo più di tipo infiammatorio o neoplastico, od anche a malformazioni congenite a carico dei tessuti cerebrali dei vasi endocranjci o delle strutture osteo-articolari craniocervicali. Nella massima parte dei casi la sintomatologia audio-vestibolare appare come elemento non costante (e quasi mai di esordio) e di solito di entità minore rispetto a sintomi più eclatanti di disfunzioni di altre strutture del sistema nervoso e ciò è indipendente dal fatto che strumentalmente si possano evidenziare alterazioni vestibolari centrali in un’alta percentuale di questi pazienti (Lòppònen et al., 1992).
Anche per ragioni di spazio non riteniamo utile descrivere queste forme in modo sistematico limitandoci quindi ad alcune importanti entità cliniche che fanno “eccezione” al comportamento a cui abbiamo sopra accennato: su di esse ci soffermeremo brevemente riportandone le caratteristiche cliniche essenziali e soprattutto gli aspetti di interesse otoneurologico.
Prima di passare alla descrizione di queste eccezioni, è comunque opportuno fare un breve inciso per richiamare l’attenzione su due patologie della fossa cranica posteriore, il neurinoma del nervo vestibolare e le malformazioni dell’arteria basilare (dolicoectasia, megabasilare, aneurismi), che prevedono l’idrocefalo tra le loro possibili complicazioni. Il neurinoma dell’acustico (per la cui descrizione si rinvia all’apposito capitolo di questo volume), pur avendo origine nel sistema nervoso periferico può nella sua evoluzione compromettere, come è ben noto, strutture nervose vicine (ponte, cervelletto, altri nervi cranici) inducendo in esse condizioni di disfunzione che, unitamente ai segni audiovestibolari, costituiscono il complesso della sindrome dell’angolo ponto-cerebellare. Questa neoplasia è peraltro causa molto frequente anche di un idrocefalo da mancato riassorbimento del liquo1 che è fortemente alterato per la presenza di un’alta quantità di proteine (Rogg et al., 2005). Questo tumore neuro periferico, attraverso la complicazione suddetta, può quindi interferire sul quadro sintomatologico abitualmente atteso condizionando eventualmente anche la presenza e/o le caratteristiche di segni e sintomi di tipo vestibolare centrale.
1’altra eccezione a cui abbiamo accennato è costituita dalle malformazioni dell’arteria basilare e tra esse soprattutto e la dolicoectasia e gli aneurismi giganti. L’aumento consistente del volume del letto vascolare e la tortuosità dell’arteria basilare sono causa di disfunzione, spesso multipla a carico di nervi cranici mentre più raramente queste alterazioni congenite ed acquisite dell’arteria basilare possono determinare ischemie a carico del tronco encefalico (Kwon et al., 2009) o condizioni di idrocefalo occlusivo per lo più attraverso la compressione sul terzo ventricolo a livello dell’acquedotto di Silvio (Kansal et al., 2011) o l’ostruzione dei forami di Monroe (Celik et al., 2012).
I sintomi relativi alla compressione sui nervi cranici sono i più precoci e frequenti (Yu et al., 1982) e possono esprimersi con nevralgie trigeminali in caso di compressione sul V nervo cranico o con alterazioni a carico dei muscoli mimici (spasmi o paresi) nell’interessamento del n. faciale, sindromi vertiginose e deficit uditivi di tipo percettivo da compromissione dell’VIli nervo; successivamente compaiono i sintomi generali dell’ ipertensione endocranica (cefalea, nausea, vomito e deterioramento cognitivo) e quelli di sofferenza focale delle strutture del tronco encefalico come atassia, vertigini, diplopia, alterazioni motorie piramidali ecc. Con discreta frequenza si riscontrano inoltre acufeni per lo più di tipo pulsante (Hafeez et al., 1999). Tra i reperti dell’esame vestibolare viene segnalata pressoché costantemente l’esistenza di un nistagmo verticale verso il basso (Jacobson e Corbett, 1989).
Tumore della fossa posteriore
Bambino di 18 mesi.
Torcicollo acquisito permanente dall'età di 13 mesi.
Difficoltà alla deambulazione (deambulazione acquisita a 12 mesi).
Segni neurologici: non vertigini ma instabilità.
Cofosi sinistra, nistagmo spontaneo sinistro, test di Halmagyi) + a destra.
Negligenza visiva dell'emicampo destro (visibile con l'inseguimento oculare e l'esame optocinetico).
Segno di Babinski + sui due lati, riflessi osteotendinei molto vivaci agli arti inferiori.
Piccola dismetria degli arti superiori.
Iporeflessia destra al test calorico e preponderanza direzionale sinistra.
TC e risonanza magnetica: astrocitoma del cervelletto.
Terapia: chirurgia (X3), radioterapia e chemioterapia.
L’atassia spino-cerebellare di Friedreich (FRDA) CASANI
È l’atassia ereditaria più comune. È una malattia neurodegenerativa autosomica recessiva con una prevalenza stimata di 1:50000-1: 29000. L’incidenza è molto più bassa negli asiatici e nei soggetti di discendenza africana. La percentuale dei portatori è stata stimata a 1:120-1:60. La malattia è causata dall’espansione ripetuta di un trinucleotide, cioè dall’espansione di una tripletta di GÀA localizzata all’interno del primo introne del gene della fratassina nel cromosoma 9q. C’è una correlazione chiara tra dimensione della ripetizione espansa e la gravità del fenotipo in FRDÀ. La fratassina è una proteina mitocondriale che ha un ruolo nell’omeostasi del ferro La deficienza di frarassina dà luogo ad accumulo mitocondriale di ferro, a difetti a carico di enzimi mitocondriali specifici, ad aumentata sensibilità agli stress ossidativi ed infine alla morte cellulare mediata da radicali liberi Le lesioni sono rappresentate da fenomeni di tipo degenerativo a carico dei cordoni posteriori e laterali del midollo spinale, delle strutture del tronco encefalico compresi i nuclei vestibolari e della corteccia cerebellare.
L’esordio di solito verso la pubertà, ma può variare da 2-3 anni di età a oltre 2.5 anni di età e solitamente è rappresentato da un disturbo della deambulazione di tipo atassico, che progressivamente si aggrava sino alla perdita della deambulazione che avviene mediamente dai 7 ai 15 anni dall’esordio della malattia e si va ad accompagnare ad altri sintomi e segni neurologici che in fase conclamata sono: riflessi tendinei assenti alle estremità inferiori, evidenza elettrofisiologica di neuropatia assonale sensitiva seguita da (entro 5 anni dall’esordio) disartria, areflessia in tutti i quattro arti, perdita distale della sensibilità di posizione e pallestesica, amiotrofia dei piccoli muscoli della mano e dei mu scoli distali della gamba e del piede, risposta piantare in estensione. Più della metà dei pazienti manifesta scoliosi progressiva, e circa la metà manifesta piede cavo, piede equinovaro e dita dei piedi ad artiglio. Altri reperti neurologici variabili includono anormalità oculomotorie, nistagmo, atrofia ottica (25%) e perdita d’udito neurosensoriale (20%).
L’anomalia del movimento oculare più comune è l’instabilità nello sguardo fisso. Non si osserva oftalmoparesi. Circa il 10% dei pazienti FRDA sono diabetici e la maggior parte di essi richiede terapia insulinica. In circa i due terzi dei pazienti vi sono alterazioni cardiache (cardiomiopatia ipertrofica) che costituiscono la causa più comune di morte che per tale patologia può verificarsi anche in età giovanile.
Le alterazioni di competenza otoneurologica sono costituite in circa la metà dei casi dalla presenza di un nistagmo spontaneo orizzontale (più raramente verticale verso il basso) o di tipo “alternante”. Nella maggior parte dei casi la reflettività vestibolare risulta ridotta. Oltre alla diminuzione dei valori dei parametri quantitativi, il nistagmo provocato dallo stimolo calorico presenta varie alterazioni qualitative (Eh et al.,1984). Il sintomo vertigine è riferito raramente ed in genere è di modesta entità, mentre sono rilevanti i disturbi della postura che si aggravano nettamente con la chiusura degli occhi.
La FRDA è una malattia a carattere progressivo con esito infausto per la quale non esistono terapie farmacologiche efficaci ma solo provvedimenti di tipo sintomatico, Questo motivo, unitamente al fatto che esistono moltissimi quadri cimici che, per lo meno in fase di esordio, possono presentarsi con una sintomatologia atassica sostanzialmente sovrapponibile a quella della malattia di Friedreich, rende indispensabile una diagnostica differenziale particolarmente scrupolosa al fine di escludere di trovarsi di fronte ad una delle cosiddette “atassie trattabili” e tra queste soprattutto l’atassia da deficit isolato di vitamina E (AVED). Si tratta di un’atassia ereditaria di origine genetica (cromosoma 8Q13, gene codificante il trasportatore dell’alfa tocoferolo) ad esordio giovanile i cui sintomi sono molto simili a quelli dell’atassia di Friedreich ma, per la quale, a differenza di questa, per lei esiste in realtà un trattamento curativo in assenza del quale le lesioni neurologiche (e la sintomatologia) continuano e si aggravano irrimediabilmente in modo progressivo.
L’esame dei livelli plasmatici di vitamina E è di conseguenza decisivo Ct la diagnosi, per l’instaurazione della terapia sostitutiva e quindi per la prognosi. Oltre alla rara AVED esistono diverse altre affezioni “trattabili” che debbono essere prese in considerazione di fronte ad una sindrome atassica e, limitandoci per motivi di spazio solo alla loro citazione, ricordiamo principalmente l’abetalipoproteinemia, la celiachia, la malattia di Refsum, il morbo di Wilson, la malattia di Wernicke-Korsakoff, la tabe, la pseudotabe alcolica, la mielosi funicolare da deficit di vitamina B12, la carenza di acido folico, la xantomatosi cerebro-tendinea, la malattia di Tay-Sachs ad esordio tardivo, la malattia di Behet, la malattia di Fabry, l’ipotiroidismo, le sindromi paraneoplastiche, le intossicazioni da ossido di carbonio e da farmaci (litio, difenilidantoina ecc.).
CAUSE DI DISFUNZIONE VESTIBOLARE
L'interconnessione del sistema vestibolare con molti altri sistemi del corpo può provocare disfunzione vestibolare secondaria ad una gamma di condizioni mediche e storie. Storie a volte associata a disfunzione del sistema periferico o centrale comprendono
· Otiti croniche o otite media Wiener-Vacher 1998/9
· Sordità congenita neurosensoriale Wiener-Vacher S.R., Toupet F. 1997 1; Amanou et al.,2000 ; Abadie et al.,2000 ; Bucci et al.,2003.
· Citomegalovirus e altre infezioni virali come nella sindrome di Ramsay Hunt (un'infezione dei nervi facciali e cocleovestibolari causata dal virus herpes zoster, lo stesso virus che è associato con la varicella)
· Malformazione da condizioni acquisite o genetiche dell'orecchio interno (malformazione o displasia di Mondini ,la sindrome Branchio-oto-renale e la sindrome di Waardenburg)
Malformazione o Displasia di Mondini
Un tale quadro può anche essere il modo di rivelarsi di una malformazione dell'orecchio interno (malformazione di Mondini) scompensata in occasione di un trauma cranico anche lieve e rivelata allora dalla TC. [11, 12, 13] Il quadro è quello di un deficit vestibolare, spesso associato a un'ipoacusia dal lato del deficit, comparso in seguito a un trauma cranico che può essere leggero o senza trauma evidente, ma senza contesto infettivo virale che precede da qualche giorno a 1 settimana (il che sarebbe piuttosto suggestivo di neurite), né otite media acuta (che sarebbe piuttosto suggestiva di labirintite). Il deficit vestibolare e la sordità sono attestati con le indagini audiovestibolari, e la TC consente di porre la diagnosi. La malformazione dell'orecchio è molto spesso isolata, a volte bilaterale, talvolta inclusa nel quadro di una sindrome malformativa più generale (sindrome di Pendred con ipotiroidismo, malformazioni vertebrali, sindrome di CHARGE che associa coloboma retinico, malformazioni cardiache, atresia coanale, ritardo dello sviluppo psicomotorio e somatico, alterazioni genitali). Le malformazioni dell'orecchio interno sono di grado variabile per la parte cocleare, ma pressoché nel 100% dei casi esiste un'assenza di canali semicircolari a livello del vestibolo. I bambini portatori di malformazioni dell'orecchio interno devono essere seguiti regolarmente, sia sul piano uditivo che vestibolare, poiché i deficit uditivi e vestibolari sono in questi casi spesso evolutivi e richiedono una gestione specifica.
· Altre malattie genetiche come la sindrome di Usher di tipo I (con grave profonda perdita neurosensoriale dell'udito e problemi di equilibrio e la visione che s deteriora dai 10 anni) o di tipo III (con problemi di equilibrio e di visione che appaiono più tardi nella vita)
Altre diagnosi
Alle vertigini possono anche essere associati segni neurologici nel quadro di malattie del sistema nervoso centrale come quelle delle intossicazioni da farmaci, delle encefaliti, delle sindromi paraneoplastiche (piuttosto frequenti con i neuroblastomi). Tuttavia, in questi casi le vertigini sono piuttosto dei disturbi dell'equilibrio piuttosto che delle reali sensazioni vertiginose e non sono in primo piano; i disturbi neurologici sono numerosi, così come ricco è il contesto clinico.
Le altre diagnosi da ipotizzare nel bambino sono più raramente riscontrate.
Le otiti croniche (in particolare colesteatomatose) possono raggiungere il labirinto posteriore creando una fistola e/o una labirintite. Questa otite cronica può essere misconosciuta e rappresentare la modalità di presentazione dell'otite. L'esame otoscopico mette in genere in evidenza una perforazione timpanica infetta o un tappo epidermico che ostruisce il condotto o una sacca di retrazione timpanica profonda e, molto più raramente (in caso di colesteatoma congenito), il timpano può essere normale o bianco, sospinto o invaso dal colesteatoma, ed è la TC delle rocche che consente la diagnosi. Il trattamento è chirurgico e deve essere intrapreso dopo una valutazione completa cocleovestibolare.
I disturbi dell'equilibrio di origine psichiatrica sono rari e soprattutto riscontrati nei bambini verso l'età di 8-10 anni. Essi sono facilmente riconoscibili per la loro atipia (sono una caricatura esagerata dei disturbi dell'equilibrio senza rapporto con condizioni di equilibrio) e per l'assenza di disturbi dell'equilibrio in alcuni comportamenti automatici (come l'allacciarsi le scarpe, la raccolta di un oggetto che è appena caduto, un cambiamento brusco non anticipato di direzione ecc.). L'esame vestibolare e neurologico è normale e un colloquio psicologico con il bambino e i genitori mette in luce un problema sottostante (aggressione a scuola, conflitti familiari...).
L'ipotensione ortostatica è soprattutto riscontrata nei bambini in periodo di crescita rapida (prepuberale e puberale) e corrisponde a un difficile adattamento transitorio del sistema cardiovascolare alle improvvise variazioni di posizione. Le vertigini compaiono in effetti in occasione dell'alzarsi al mattino (passaggio troppo brusca dalla posizione sdraiata alla posizione eretta) o durante la stazione eretta prolungata. Le sensazioni di vertigine si accompagnano a sensazione di testa vuota, fosfeni e cefalee e durano da alcuni secondi a qualche minuto. Esse possono anche essere accompagnate da lipotimie, raramente da perdita di coscienza. L'esame vestibolare è normale e la diagnosi è confermata con misurazione della pressione arteriosa in posizione supina dopo 20 minuti di decubito dorsale e al momento della brusca assunzione della posizione eretta. I valori pressori della massima cadono di 20-40 mmHg al momento della posizione eretta mentre la manovra riproduce le sensazioni di cui si lamenta il giovane paziente. Il trattamento consiste nell'evitare i cambiamenti improvvisi di posizione.
La vertigine parossistica benigna posizionale (VPPB) in rapporto con una patologia otolitica (migrazione di cristalli otolitici nei dotti oppure sulla cupola dei canali semi-circolari) è soprattutto una patologia dell'adulto. Queste vertigini posizionali si manifestano raramente nel bambino e in genere piuttosto in un contesto traumatico (trauma cranico in seguito a una caduta o un incidente stradale). Le caratteristiche sono le stesse che nell'adulto: vertigine che compare al momento di un cambiamento di posizione con nistagmo geotropico con una latenza, estinguibile, adattabile. La sensazione vertiginosa è spesso meglio tollerata dai bambini che dagli adulti. Le valutazioni otologica, vestibolare e neurologica sono normali, il trattamento comporta come per gli adulti una manovra liberatoria (Semont-Toupet, Epley).
La malattia di Ménière e le sindromi di Ménière sono rare nel bambino e, nella nostra esperienza, le sindromi di Ménière che associano vertigini i, ipoacusia e acufeni con sensazione di orecchio ovattato sono state osservate solo a partire dall'età di 2-8 anni. Forse è una diagnosi sottostimata poiché i sintomi non sono sempre ritrovati all'anamnesi e, per esempio, l'ipoacusia (predominante sulle frequenze gravi) può essere una scoperta occasionale all'audiogramma richiesto in presenza di vertigini anche quando il bambino non aveva avvertito questa ipoacusia. Gli acufeni non sono necessariamente riferiti spontaneamente dal bambino. La vertigine è di regola il sintomo principale, invalidante, associata a nausea e vomito, senza cefalea e della durata di diverse ore o anche di una giornata. Se il bambino è visitato nel corso della crisi, si può constatare il nistagmo spontaneo spesso intenso. Gli esami otologico, neurologico sono normali e l'esame vestibolare al di fuori delle crisi è il più delle volte normale. L'audiogramma dimostra la caduta delle soglie uditive sulle frequenze gravi mentre la timpanometria esclude un problema tubarico. La misura dei PEOM può aiutare nella diagnosi mostrando un'assenza di risposta sul lato della sordità (in conduzione aerea e in conduzione ossea). Il test al glicerolo (esame molto lungo con due audiogrammi a 1 ora di intervallo) è raramente realizzato nel bambino, ma può mostrare un miglioramento delle soglie uditive 1 ora dopo l'assunzione di glicerolo, che indica la presenza di un'idrope. Questo tipo di test non è tuttavia sempre positivo. Bisogna guardarsi dal porre questa diagnosi al momento di una prima crisi, controllare il bambino regolarmente ed essere pronti a rimettere in causa questa diagnosi se la sintomatologia cambia. Se le vertigini sono ripetute e invalidanti, si può prescrivere una terapia di lunga durata con betaistina, che non ha alcun effetto secondario nel bambino.
La delayed vertigo o vertigine ritardata si verifica in genere da alcuni mesi a qualche anno dopo una lesione dell'orecchio interno con una sordità secondaria a un trauma (esempio: frattura della rocca) o una lesione infettiva o virale (esempio: la parotite prima della vaccinazione morbillo-parotite-rosolia [MPR] era una malattia che spesso produceva cifosi e delayed vertigo; la frequenza delle delayed vertigo è peraltro diminuita in modo notevole da quando esiste la vaccinazione sistematica dei bambini). Queste delayed vertigo possono manifestarsi come delle sindromi di Ménière ma nella storia del paziente è presente la nozione di una lesione dell'orecchio interno e la sindrome è spesso incompleta, senza segni uditivi associati alla crisi vertiginosa. Il meccanismo evocato all'origine di queste vertigini sarebbe molto vicino a quello evocato nelle sindromi di Ménière e i trattamenti delle delayed vertigo identici a quelli proposti nella malattia di Ménière. Quando le vertigini resistono alla terapia medica, si propone più facilmente, in caso di cofosi, una labirintectomia chirurgica o chimica.
Le vertigini d'origine comiziale sono di regola rare e fanno parte delle aure delle crisi comiziali, generalmente associate a un corteo di segni che evocano una comizialità: allucinazioni di movimenti complessi (più che una vertigine rotatoria isolata), allucinazioni uditive, segni neurologici di localizzazione, assenza con perdita di conoscenza. Un tale quadro impone una consulenza neurologica che sarà in genere seguita da un elettroencefalogramma e un'indagine diagnostica cerebrale.
La lesione auto-immune dell'orecchio interno, [Ndiaye et al.,2002] o sindrome di Cogan, si manifesta con una "banale congiuntivite". Nei tre casi osservati in 12 anni, ogni volta la sindrome infiammatoria è stata secondaria a una stimolazione eccessiva del sistema immunitario (BCGite dopo una seconda vaccinazione BCG, parotite dopo un richiamo di vaccinazione MPR in un ragazzo di 14 anni, dopo le prime inoculazioni di desensibilizzazione contro gli acari). Il bambino ha gli occhi rossi e ciò è interpretato come una congiuntivite virale, ma svilupperà una sordità improvvisa con instabilità o vertigine che segna il danno vestibolare talvolta progressivo. La valutazione laboratoristica mostra una sindrome infiammatoria importante e l'esame oftalmologico una cheratite interstiziale evocatrice. È una diagnosi rara ma deve essere conosciuta perché deve essere prescritto un trattamento steroideo a forti dosi in urgenza per evitare la perdita dell'udito.
Nelle vertigini del bambino, bisogna dunque sempre effettuare un esame clinico otologico, neurologico e vestibolare (comprendente in particolare il test di Halmagyi di rotazione rapida della testa e la ricerca di un nistagmo spontaneo e provocato sotto occhiali di Frenzel o da videoscopia) prima di precipitarsi su esami complementari eccessivi e costosi. I primi esami diagnostici da richiedere in priorità sono un esame vestibolare (che comprende almeno un test calorico e dei PEOM) e un esame oftalmologico per escludere un disturbo della rifrazione e un disturbo della vergenza od oculomotorio. Quando ricorrere alla diagnostica per immagini? Certamente non di prima intenzione, tranne il caso in cui l'esame neurologico mostri delle anomalie che possono fare sospettare un processo espansivo endocranico o se un contesto di trauma cranico si associa a segni che evocano la possibilità di una frattura della rocca. La TC sarà allora il primo esame da richiedere in quanto più rapida (meno di 1 min di immobilizzazione richiesta con le nuove tecniche di TC elicoidale). La RM potrà essere realizzata per precisare la natura delle immagini individuate alla TC. Nel bambino giovane un esame diagnostico (RM o TC) richiederà una premedicazione, o anche un'anestesia generale, e questi esami rappresentano dunque un rischio non trascurabile; inoltre sono molto costosi. Non è dunque giustificato prescrivere questi esami di fronte a una vertigine nel bambino prima di aver effettuato un esame clinico oto-neuro-vestibolare completo, una valutazione audiovestibolare e una visita oftalmologica.
Riferimenti bibliografici
Abadie V., Wiener-Vacher S.R., Morisseau-Durand M.P., Poree C., Amiel J., Amanou L., e al. Vestibular anomalies in CHARGE syndrome: investigations and consequences for postural development Eur. J. Pediatr. 2000 ; 159 : 165-171
Agrup C. Immune-mediated audiovestibulari disorders in the pediatric population: a review. Int J Aud. 2008;47:56–565.
Akin FW, & Murnane, OD (2000). Vestibular evoked
myogenic potentials: preliminary report. Journal of the American
Academy of Audiology, 12 (9), 445-52.
Andrieu-Guitrancourt, JA, Peron, JM, Deeside, D., Aubet, J., & Courtin, P.
(1981). Normal vestibular responses to air caloric tests in children. ,
245-250.
Amanou L., Morisseau-Durand M.P.,
Wiener-Vacher S.R., Marianovski R., Abadie V., Manac'h Y.
L'oreille interne dans l'association CHARGE Ann. Otolaryngol. Chir.
Cervicofac. 2000 ; 117 : 161-167
Angeli S. Value of vestibular testing in young children with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg. 2003; 129(4):478–482.
Anoh-Tanon M.J., Bremond-Gignac D., Wiener-Vacher S.R. Vertigo is an underestimated symptom of ocular disorders: dizzy children do not always need MRI Pediatr. Neurol. 2000 ; 23 : 49-53 [cross-ref]
Bacher E, Wright C, Karmody C. The incidence and distribution of cupular deposit in the pediatric vestibular labyrinth. Laryngoscope. 2002;112:147–151.
Balkany T, Finkel R. The dizzy child. Ear Hear. 1986; 7(3):138–142.
Biel L. Raising a sensory smart child. Penguin Books. 2009.
Batson G. Benign paroxysmal vertigo of childhood: a review of the literature. Ped Child Health. 2004; 9:31–34.
Bauer F, Westhofen M. Vestibulotoxic effects of the cytostatic drug carboplatin in patient with head and neck tumors. HNO. 1992; 40:19–24.
Brookhouser, PE, Cyr, DB, & Beauchaine, KA (1982). Vestibular findings in the deaf and hard of hearing. Otolaryngology: Head and Neck Surgery, 90 , 773-777
Braswell J, Rine R. Evidence that vestibular hypofunction affects reading acuity in children. Int J Pediat Otorhinolaryngol. 2006; 70(11):1957–
1965.
Bucci M.P., Kapoula Z., Yang Q., Wiener-Vacher S., Bremond-Gignac D. Saccades, vergence and combined movements in a young subject with Congenital Central Hypoventilation Syndrome (CCHS) Strabismus 2003 ; 11 : 95-107 [cross-ref]
Bucci M.P., Kapoula Z., Yang Q., Wiener-Vacher S., Bremond-Gignac D. Abnormalities of vergence latency in children with vertigo J. Neurol. 2004 ; 251 : 204-213 [cross-ref]
Bucci M.P., Kapoula Z., Yang Q., Bremond-Gignac D., Wiener-Vacher S. Speed-accuracy of saccades, vergence and combined movements in children with vertigo Exp. Brain Res. 2004 ; 157 : 286-295
Casselbrant M, Villardo R, Mandel E. Balance and otitis media with effusion.Int J Audiol. 2008; 47(9):584–589.
Colebatch, JG,
& Halmagyi, GM (1992). Vestibular evoked potentials in human neck
muscles before and after unilateral vestibular deafferentation. Neurology,
42 , 1635-1636.
Chang C, Young Y. Caloric and vestibular evoked myogenic potential
tests in evaluating children with benign paroxysmal vertigo. Int J Pediatr
Otorhinolaryngol. 2007;71(3):495–499Church M, Gerkin K. Hearing disorders in children with fetal alcohol syndrome: findings from case reports. Pediatrics.1988; 82:147–154.
Cyr, DG (1980). Vestibular testing in children. Annals of Otology, Rhinology and Laryngology, 89 , 63-69.
Cyr, DG
(1983). The vestibular system: pediatric considerations. Seminars
in Hearing, 4 (1), 33-45.
Cyr, DG, Moore GF, & Moller, CG (1988). Clinical application of
computerized dynamic posturography. Entechnology, September ,
36-47.
Di Fabio RP & Foudriat PA (1995). Responsiveness and reliability of a pediatric strategy score for balance. Physiology Research International, 1 (3), 180-194.
Donat, JFG, Donat, JR, & Swe Lay, K. (1980). Changing response to caloric stimulation with gestational age in infants. Neurology, 30 , 776-778.
Enbom H, Magnusson M, Pyykko I. Postural compensation in children with congenital or early acquired bilateral vestibular loss. Ann Otol Rhinol
Laryngol. 1991; 100(6): 472–478.
Eviatar, L., Eviatar, A., & Naray, I. (1974). Maturation of neuro vestibular responses in infants. Developmental Medical Child Neurology, 16 , 435-446.
Eviatar,
L., Miranda, S., Eviatar, A., Freeman, K., & Borkowski, M. (1978). Development
of nystagmus in response to vestibular stimulation in infants. Annals
of Neurology, 5 , 508-514.
Fife, TD, Tusa, RJ, Furman, JM, Zee, DS, Frohman, E., Baloh, RW, Hain, T., Goebel,
J., Demer, J., & Eviatar, L. (2000). Assessment: vestibular testing
techniques in adults and children. Neurology, 55 ,
1431-1441.
Flechsig P (1920) Anatomie des menschen Gehirns und Rückenmarksauf Myelogenetischer Grudlage. Georg Theme, Leipzig
Frick S, Kawar M. Vestibular habituation from the core: Clinical reasoning,© Vestibular Disorders Association ◦ www.vestibular.org ◦ Page 8 of 10 assessment, and intervention skills through enhanced understanding of the interaction within the vestibularauditory-visual triad. Vital Links.www.VitalLinks.net.
Gagnon I, Swaine B, Friedman D, et al.Children show decreased dynamic balance after mild traumatic brain injury. Arch Phys Med Rehabil. 2004;85(3):444–452.
Halmagyi, GM, & Colebatch, JG (1995). Vestibular evoked myogenic potentials in the sternocleidomastoid muscle are not of lateral canal origin. Acta Otolarngologica Supplement, 520 (1), 1-3.
Hirabayashi, S. & Iwasaki, Y. (1995). Developmental perspective of sensory organization on postural control. Brain Development, 17 (2), 111-113.
Hullar TE, Zee DS, Minor LB, Evaluation of the patient with dizziness, in: Harker LA, ed. Cummings Otolaryngology Head & Neck Surgery. VoI Four Philadelphia: Elsevier Mosby 2005: 3160—3198
Jinkowic T, Gelo J, Astley S. Children and youth with fetal alcohol spectrum disorders: summary of intervention and recommendations after clinical diagnosing. Intellect Dev Disabil. 2010;48:330–344.Kaga K (1994) Structure of vertigo. Kanehara Shuppan, Tokyo
Kaga K, Sakurai H, Ogawa Y, Mizutani T, Toriyama M (2001) Morphological changers of vestibular ganglion cells in human fetuses and in pediatric patients. Int J Pediatr Otorhinolaryngol 60:11–20
Kamoshita S (1972). Development of neurological system. Progression of pediatric neurology, vol 1. Diagnosis and Treatment, Tokyo
Keating, N. A comparison of duration of nystagmus as measured by the Southern California Post-rotary Nystagmus Test and electronystagmography. Amer J Occ Ther. 1979; 33:92–96.
Kelsch, TA,
Schaefer, LA, & Esquivel, CR (2006). Vestibular evoked myogenic
potentials in young children: test parameters and normative data. Laryngoscope,
116 (6), 895-900.
Kenyon, GS (1988). Neuro-otological findings in normal children. Journal
of the Royal Society of Medicine, 81 , 645-648.
Krejcova, H., Filipova, M., & Krejci,
L. (1975). Vestibulometric evaluation of the caloric reaction in
children and in adults. Electrophysiology Clinical Neurophysiology, 39 (4),
441.
Levens, SL (1988). Electronystagmography in normal children. British
Journal of Audiology, 22 , 51-56.
Lightfoot G, Barker F, Belcher K, Kennedy V, Nassar G, Tweedy F. The derivation of optimum criteria for use in the monothermal caloric screening test. Ear Hear . 2009;30(1):54-62.
Marcelli V, Piazza F, Pisani F, Marciano E. Neuro-ontological features of benign paroxysmal vertigo and benign paroxysmal positioning vertigo in children. Brain Devel. 2006; 28:80–84.
Melagrana A, D'Agostino R, Tarantino V, Taborelli G, Calevo MG. Monothermal air caloric test in children. Int J Ped Otorhinolaryngol . 2002;62(1):11-15.
Mulch, G. & Peterman, W. (1979). Influence of age on results of vestibular function tests: review of literature and presentation of caloric test results. Annals of Otology, Rhinology, and Laryngology, 88 , 1-17.
Murnane OD, Akin FW, Lynn SG, Cyr DG. Monothermal caloric screening test performance: a relative operating characteristic curve analysis. Ear Hear . 2009;30(3):313-319.Nandi R, Luxon LM. Development and assessment of the vestibular system. Int J Audiol . 2008;47:566-577.
Ndiaye I.C., Rassi S.J., Wiener-Vacher S.R. Cochleovestibular impairment in pediatric Cogan's syndrome Pediatrics 2002 ; 109 : E38
Ornitz, EM, Atwell,
CW, Walter, DO, Hartmann, EE, & Kaplan, AR (1979). The maturation of
vestibular nystagmus in infancy and childhood. Acta Otolaryngologica,
88 , 244-256.
Niemensivu R, Kentala E, Wiener-Vacher S, Pyykko I. Evaluation of
vertiginous © Vestibular Disorders Association ◦ www.vestibular.org
◦ Page 9 of 10 children. Eur Arch Otorhinolaryngol.2007;
264(10):1129–1135.
Rine R, Rubish K, Feeney C. Measurement of sensory system effectiveness and maturational changes in postural control in young children. Ped Phys Ther. 1998;10:16–22.
Rine R, Cornwall G, Gan K, et al. Evidence of progressive delay of motor development in children with sensorineural hearing loss and concurrent vestibular dysfunction.
Percept Mot Skills. 2000; 90:1101–1112.
Rine, RM, Cornwal,l
G., Gan, K., LoCascio, C., O-Hare, T., Robinson, E., & Rice,
M.(2000). Evidence of progressive delay of motor development in children
with sensorineural hearing loss and concurrent vestibular dysfunction. Perceptual
Motor Skills, 90 , 1101-1112.
Rine R, Braswell J. A clinical test of dynamic visual acuity for
children. Int J Ped Otorhinolaryngol. 2003;67(11):1195–1201.
Rine R, Braswell J, Fisher D, et al.Improvement of motor development and postural control following intervention in children with sensorineural hearing loss and vestibular impairment.Int J Ped Otorhinolaryngol. 2004;68(9):1141–1148.
Rine RM. Growing evidence for balance and vestibular problems in children.Audiol Med. 2009; 7(3):138–142.Sheykholeslami, K., Megerian, CA, Arnold, JE, & Kaga, K. (2005). Vestibular evoked myogenic potentials in infancy and early childhood. Laryngoscope, 115 (8), 1440-1444.
Shimizu, K., Asai, M., Takata, S., & Watanabe, Y. (1994). The development of equilibrium function in childhood. In: K. Taguchi, M. Igarashi, & S. Mori, (Eds.), Vestibular and Neural Front (pp. 183-186). New York, NY: Elsevier Science BV
Snashall SE. Vestibular function tests in children. J Royal Soc Med . 1983;76:555-559.Suarez H, Angeli S, Suarez A, et al. Balance sensory organization in children with profound hearing loss and cochlear implants. Int J Pediatr Otorhinolaryngol.2007; 71(4):629–637.
Staller, SJ, Goin, DW, & Hildebrandt, M. (1986). Pediatric vestibular evaluation with harmonic acceleration. Otolaryngology Head & Neck Surgery, 95 (4), 471-476. Uneri A, Turkdogan D. Evaluation of vestibular functions in children with vertigo attacks. Arch Dis Child. 2003;68:510–511.
Valente, M. (2007). Maturational effects of the vestibular system: a study of rotary chair, computerized dynamic posturography and vestibular evoked myogenic potentials with children. Journal of the American Academy of Audiology, June 18 (6), 461-481.
Van der Laan, FL, & Oosterveld, WJ (1974). Age and vestibular function. Aero Medicine, 45 (5), 540-547.
Viccaro M, Mancini P, La Gamma R, et al. Positional vertigo and cochlear implantation. Otol Neurotol. 2007;28:764–767.
Waldron M, Matthews J, Johnson I. The effect of otitis media with effusions on balance in children. Clin Otolaryngol Allied Sci. 2004; 29:318–320.
Weiss A, Phillips J. Congenital and compensated vestibular dysfunction in childhood: An overlooked entity. J Child Neurol. 3. 2006; 21(7):572–579.
Rine RM. Growing evidence for balance and vestibular problems in children. Aud Med 2009; 7:138–142.
Wiener-Vacher S.R., Toupet F. Quelle est la place de l'appareil vestibulaire dans les troubles de l'équilibre de l'enfant? Évol Psychomotrices 1997 ; 9 : 212-215
Wiener-Vacher S.R., Toupet F., François M., Van Den Abbeele T., Viala P., e al. Conséquences des déficits vestibulaires sur le développement du contrôle postural chez l'enfant Journées parisiennes de pédiatrie Paris: Flammarion (1998). 283-287
Wiener-Vacher S.R. Conséquences posturo-locomotrices de déficits vestibulaires chez l'enfant. Posture et équilibre Marseille: Solal (2001). p. 87–94.
Wiener-Vacher S.R., Bril B., Ledebt A. Changes in otolithic function in infants learning to walk observed with off vertical axis rotation Neuroscience 1994 ; 20 : 1108[abstract].
Wiener-Vacher S.R., Toupet F., Narcy P. Canal and otolith vestibulo-ocular reflexes to vertical and off vertical axis rotation in children learning to walk Acta Otolaryngol. 1996 ; 116 : 657-665
Wiener-Vacher S.R., Ledebt A., Bril B. Changes in otolith VOR to Off vertical axis rotation in infants learning to walk: preliminary results of a longitudinal study Ann. N. Y. Acad. Sci. 1996 ; 781 : 709-712 [cross-ref]
Wiener-Vacher S.R., Toupet F., Elmaleh M., Narcy P. Pathologie cochléo-vestibulaire post-traumatique chez l'enfant Lettre ORL Chir. Cervicofac 1998 ; 236 : 21-23: 88-97
Wiener-Vacher S.R., Amanou L., Denise P., Narcy P., Manach Y. Vestibular function in children with CHARGE association Arch. Otolaryngol. Head Neck Surg. 1999 ; 125 : 342-347
Wiener-Vacher S.R. What is useful in vestibular testing Otorhinolaryngol Nova 2001 ; 11 : 95-98 [cross-ref]
Wiener-Vacher S.R. Clinical application of the Off Vertical Axis Rotation test (OVAR) Adv. Otorhinolaryngol. 2001 ; 58
Weiner-Vacher S. Vestibular disorders in children. Int J Aud. 2008; 47:578–583.
Steenerson R, Cronin G, Gary L. Vertigo after cochlear implantation. Otol Neurotol. 2001; 22(6):842–843.
Worden B, Blevins N. Pediatric vestibulopathy and pseudovestibulopathy: differential
diagnosis and management. Curr Opin Otolaryngol Head Neck Surg. 2007;15(5):304–309
Yukawa K, Hagiwara A, Ogawa Y, et al. Bilateral progressive hearing loss and vestibular dysfunction with inner ear antibodies. Auris Nasus Larynx. 2010;37:223–228.
© 2005 Elsevier SAS. Tutti i diritti riservati.
Cause di disfunzione vestibolare
L'interconnessione del sistema vestibolare con molti altri sistemi del corpo può causare disfunzione vestibolare secondaria ad una serie di condizioni mediche e storie. Storie a volte associate a disfunzioni del sistema periferico o centrale sono:
- Sordità neurosensoriale Congenita 1,12,13,14
- Cytomegalovirus e altre infezioni virali come nella sindrome di Ramsay Hunt (un'infezione dei nervi facciali e cocleovestibolari causata dal virus herpes zoster, lo stesso virus che è associato con varicella)
- Malformazioni da condizioni acquisite o genetiche come la sindrome di branchio-otorenale, Mondini displasia, e la sindrome di Waardenburg
- Altre malattie genetiche come la sindrome di Usher di tipo I (con grave profonda perdita dell'udito neurosensoriale e problemi di equilibrio e visione deteriorata da 10 anni) o di tipo III (con problemi di equilibrio e la visione che compaiono più tardi nella vita)
- Disturbi o condizioni come la paralisi cerebrale, idrocefalo, un tumore al cervello posteriore, o sindrome Wallenberg (causata da un ictus dal blocco nel inferiore dell'arteria cerebellare vertebrale o posteriore del tronco cerebrale) Neurologiche
Sindrome branchiootorenale BOR, Sindrome BOR, Displasia branchiootorenale, Branchio-Otorenale Displasia,Branchio-Otorenale Syndrome, Sindrome Branchio-Oto-Renale, Sindrome di Melnick-Fraser
- trasmissione autosomica dominante Fig.1
- espressività variabile
- sordità neurosensoriale. mista, trasmissiva Fig.2
- bottoni preauricolari Fig.3
- cisti-fistole del collo (branchiali) Fig.4
- malformazione del padiglione auricolare Fig.5
- malformazioni renali (agenesia-ipoplasia)Fig.6
|
|
|
|
|
Fig.1
|
Fig.2 |
Fig. 3 |
|
|
|
|
|
Fig.4 |
Fig.5 |
Fig.6 |

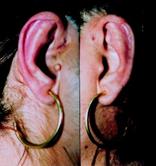
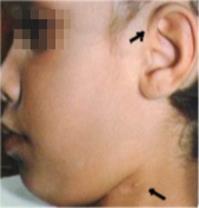
Che cosa è la sindrome Branchio-Oto-Renale?
LA Sindrome Branchio-Oto-Renale (BOR) è una condizione genetica, nota anche come sindrome di Melnick-Fraser, è caratterizzata da un'associazione di: 1) fistole brachiale o cisti lungo i lati del collo corrispondenti alla posizione delle fessure branchiali embriologiche; 2) malformazioni dell'orecchio, che possono includere l'orecchio interno, medio ed esterno; 3) malformazioni renali, che possono variare in gravità da ipoplasia renale alla agenesia con conseguente insufficienza renale. I segni ed i sintomi di questa condizione , tuttavia, possono variare.
"Branchio-" si riferisce al secondo arco branchiale, che è una struttura che negli embrioni dà origine ai tessuti nella parte anteriore e laterale del collo. Nelle persone con sindrome branchiootorenale , sviluppo anormale del secondo arco branchiale possono causare la formazione delle masse al collo chiamato cisti branchiale leporino. In alcune persone, connessioni anormali chiamati modulo fistole passaggi tra queste cisti e la superficie del collo. Fistole può anche svilupparsi tra la pelle del collo e della gola, vicino alle tonsille. Cisti schisi branchiali e fistole possono causare problemi di salute se diventano infette.
"Oto-" si riferisce all'orecchio; la maggior parte delle persone con sindrome branchiootorenale hanno perdite e altre anomalie dell'orecchio dell'udito. La perdita dell'udito è conosciuta come la sordità neurosensoriale se è causato da cambiamenti nella parte interna dell'orecchio e sordità conduttiva se è causato da cambiamenti dell'orecchio medio. La sindrome Branchio-Oto-Renale può anche comportare la perdita che deriva da cambiamenti sia l'orecchio interno e l'orecchio medio, che si chiama perdita uditiva mista dell'udito. Altre anomalie dell'orecchio associate alla sindrome di Branchio-Oto-Renale comprendono malformazioni dell'orecchio interno o l'orecchio medio e orecchio esterno di forma anomala (padiglioni auricolari). Alcune persone colpite hanno anche piccoli fori nella pelle (pit preauricolari) o piccoli lembi di pelle (tag preauricolari) proprio di fronte l'orecchio.
"Renale" si riferisce ai reni; La sindrome Branchio-Oto-Renale provoca alterazioni della struttura e della funzione renale. Queste anomalie vanno da lievi a gravi e possono interessare uno o entrambi i reni. In alcuni casi, la malattia renale allo stadio terminale (ESRD) si sviluppa più tardi nella vita. Questa grave condizione si verifica quando i reni diventano incapaci di filtrare efficacemente i liquidi ei prodotti di scarto dal corpo.
Manifestazioni generali
La displasia renale è riportato in oltre due terzi dei pazienti e varia da pali rastremati superiori (duplicazione del sistema di raccolta) alla marcata agenesia del rene. La sindrome può includere sequenza Potter.
Ereditata in modo autosomico dominante, ogni bambino di un genitore con la sindrome di BOR ha una probabilità del 50% di presentare con la malattia. Si pensa che il 90% dei casi di sindrome BOR sono dovuti a eredità, mentre il rimanente 10% dei casi sono dovuti a mutazioni acquisite. BOR mostra espressività variabile, che rappresentano le differenze nella gravità dei sintomi tra i membri della famiglia. Penetranza, tuttavia, è 100%. Attesa (propensione per le generazioni successive di presentare con la malattia in età più giovane) non si verifica.
Storia
• Nel 1864 Heusinger è stato il primo a riconoscere un'associazione tra branchiale fistole leporino, pozzi preauricolari, e compromissione dell'udito.
• Nel 1975, Melnick et al. e Fraser et al. descritto sindrome BOR come entità specifica con un pattern di ereditarietà autosomica dominante.
Epidemiologia, Quanto è diffusa la sindrome Branchio-Oto-Renale ?
I ricercatori stimano che la sindrome Branchio-Oto-Renale colpisce circa 1 su 40.000 persone.
Quali geni sono legati alla sindrome Branchio-Oto-Renale ?
Le mutazioni in tre geni, EYA1, SIX1 e SIX5, sono noti per causare la sindrome Branchio-Oto-Renale . Circa il 40 per cento delle persone con questa condizione hanno una mutazione nel gene EYA1. Eredità autosomica dominante indica che il gene difettoso responsabile di una malattia si trova in una autosoma , e solo una copia del gene è sufficiente a causare la malattia, quando ereditato da un genitore che ha il disturbo. Mutazioni SIX1 eSIX5 sono cause molto meno comuni della malattia.
Le proteine prodotte dal EYA1, SIX1, ei geni SIX5 svolgono un ruolo importante nello sviluppo prima della nascita. La proteina EYA1 interagisce con diverse altre proteine, tra cui SIX1 e SIX5, per regolare l'attività dei geni coinvolti in molti aspetti dello sviluppo embrionale. La ricerca suggerisce che queste interazioni proteine sono essenziali per la normale formazione di molti organi e tessuti, tra cui il secondo arco branchiale, orecchie e reni. Mutazioni nel EYA1, SIX1, o gene SIX5 spesso interrompono la capacità delle proteine 'di interagire uno con l'altro e regolare l'attività dei geni. Le conseguenti variazioni genetiche influenzano lo sviluppo di organi e tessuti prima della nascita, che porta gli elementi caratteristici della sindrome Branchio-Oto-Renale .
Mutazioni Eya1 e SIX1 sono stati trovati anche in alcune persone con una condizione nota come branchio-oto (BO) sindrome. Questa condizione include molte delle stesse caratteristiche come la sindrome branchiootorenale, ma individui affetti non hanno renali (renale) anomalie. Le due condizioni sono altrimenti così simili che i ricercatori spesso li considerano insieme (BOR / sindrome BO). Non è chiaro il motivo per cui alcune mutazioni Eya1 e SIX1 sono associati ad anomalie renali e altri non lo sono.
Alcune persone con la sindrome di Branchio-Oto-Renale non hanno una mutazione identificata nel EYA1, SIX1,o gene SIX5. In questi casi, la causa della condizione è sconosciuta.
Per saperne di più sul EYA1 , SIX1 , e SIX5 geni.
Come i pazienti ereditano la sindrome Branchio-Oto-Renale ?
Questa condizione è ereditata come carattere autosomico dominante, il che significa una sola copia del gene alterato in ogni cella è sufficiente a causare il disturbo. In circa il 90 per cento dei casi, una persona affetta eredita la mutazione da un genitore affetto. I casi rimanenti derivano da nuove mutazioni nel gene, e si verificano in persone senza storia di malattia nella loro famiglia.
Manifestazioni otorinolaringoiatriche
Le malformazioni auricolari comprendono pozzi preauricolari , che sono ciechi depressioni capocchia di spillo nella parte superiore del padiglione auricolare dell'orecchio. Essi si osservano in 75-85% dei pazienti. Altre malformazioni dell'orecchio esterno (40-60%) possono includere tag preauricolari, orecchie a pipistrello e microtia. I canali uditivi esterni possono essere atresici.
Anomalie dell'orecchio medio comprendono anomalie del ossicini, il nervo facciale, ei canali di Falloppio. La tomografia computerizzata delle ossa temporali può dimostrare coclea ipoplasico e canali semicircolari assenti o ipoplasia. È stata inoltre segnalata Mondini displasia. La perdita dell'udito (75-90%) di solito è stabile e può essere conduttiva, neurosensoriale o mista. Si stima che il 2% dei bambini con sordità profonda hanno la sindrome BOR.
Descritta per la prima nel 1975, da M. Melnick, e nel 1978 da FC Fraser. La sindrome di BOR (nota anche come sindrome di Melnick-Fraser) è l'associazione di malformazioni auricolari, fistole branchiali(63%) della sindrome BOR sono solitamente bilaterale e nella parte inferiore del collo con aperture esterne sul bordo mediale del muscolo sternocleidomastoideo, sordità e anomalie renali. La prevalenza è stimata in 1 caso ogni 40.000 popolazione generale. Altre manifestazioni associate comprendono l'aplasia o stenosi dei dotti lacrimali (8-9%), palato alto arcuato o spacco, morso profondo, e anomalie del nervo facciale. Alcuni autori considerano la microsomia emifacciale (HFM) per essere una forma grave. Vedere emifacciale microsomia, nella sezione Sindrome di Goldenhar. La sindrome branchio-oto-renale (BOR) associa sordità, fistole branchiali multiple e una malformazione renale. La sua prevalenza è stimata a 1/ 40.000(Fraser F.C.). Le malformazioni renali possono essere notevoli (agenesie oppure ipoplasie maggiori) e portano a volte a un'interruzione della gravidanza. Le malformazioni meno importanti saranno diagnosticate con un'ecografia renale che deve essere richiesta di fronte a una sordità suggestiva di BOR: la sordità si accompagna a malformazioni dell'orecchio esterno (malformazioni del padiglione, aplasia dell'orecchio, encondromi, stenosi dei condotti uditivi) (Fig. 4 ), dell'orecchio medio (esiste una componente di trasmissione all'audiogramma) e dell'orecchio interno (varie malformazioni cocleovestibolari) (Fig. 5 ). Si ritrovano in generale delle fistole preauricolari bilaterali e possibilità di apertura di fistole della seconda fessura branchiale con residui cartilaginei associati suggestivi (Fig. 6 ) In presenza di questi segni. un’ecografia renale può confermare questa sindrome.. In pratica, in presenza di una sordità di percezione o mista associata a una fistola branchiale oppure a malformazioni dell'orecchio esterno, è consigliabile eseguire un'ecografia renale. Tre geni sono stati localizzati e due identificati, EYA1 e SIX1(Abdelhak S., Ruf R.G.). Il gene EYA1 può anche essere responsabile di una sindrome branchio-otologica, molto simile alla BOR ma senza interessamento renale(Vincent C.).
· EYA1 (situato sul cromosoma 8 , [8q13.3]), più di 100 tipi di mutazioni responsabili di BOR, le mutazioni sono divisi in tre gruppi principali (isoforma A, B e C) (presente nel 38% -40,5% dei pazienti)
· Six1 (situato sul cromosoma 14 , [14q23.1]), almeno quattro tipi di mutazioni responsabili della BOR (rilevato in 5% dei pazienti),
· SIX5 (situato sul cromosoma 19 , [19q13.32]), almeno quattro tipi di mutazioni responsabili della BOR (rilevato in più dell'1% dei pazienti).
Eziologia e la patogenesi
La sindrome BOR viene ereditata come carattere autosomico dominante con elevata penetranza ed espressione variabile. La patogenesi si presume essere una carenza nella differenziazione del primo e secondo arco branchiale. Le anomalie del sistema renale sono il risultato dell'interazione anomala tra la gemma ureterale e il blastema metanefrico. L'orecchio interno (stria vascularis) ed i glomeruli renali condividono un antigene comune, e più di un gruppo di studio ha descritto i cambiamenti istopatologici su esami dell'osso temporale.
|
|
|
|
Fig. 4 : Orecchie a trombetta ed encondromi. |
Un bambino di 13-mesi con sintomi di BOR. L'orecchio sinistro appositamente sviluppato; all'interno l'orecchio destro, il canale uditivo e atresia lobo impropriamente sviluppati. |
|
|
|
|
Fig. 5 : Micrococlea in una sindrome branchio-oto-renale (BOR). |
|
|
|
|
|
Fig. 6 : Residuo branchiale cervicale |
Ragazza con la sindrome di BOR, fistola carotidea bilaterale |
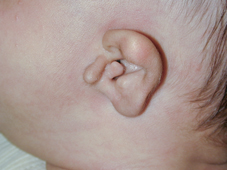
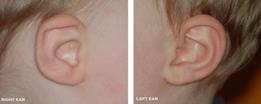
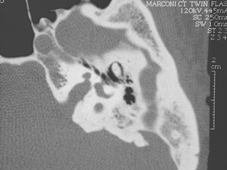
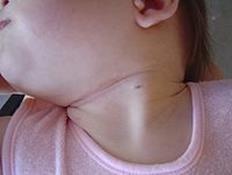
Una storia familiare positiva per brachiale, otologica, e le malformazioni renali è suggestivo di sindrome di BOR.
Se nessuna storia familiare di malattia è noto, criteri clinici possono essere utilizzate per fare la diagnosi per pazienti con almeno tre criteri principali. In alternativa, la diagnosi può essere fatta per i pazienti che dimostrano due criteri principali e due criteri minori.
Criteri principali
1. Malformazione dei padiglioni auricolari
2. Perdita dell'udito
3. Encondromi preauricolari
4. Anomalie renali
5. Anomalie secondarie dell’arco branchiale
6. Stenosi del canale uditivo esterno
Criteri minori
1. Appendici preauricolari
2. Aplasia del dotto lacrimale
3. Anomalie dell'orecchio medio
4. Anomalie dell'orecchio interno
5. Malformazioni del palato o asimmetria facciale
6. Gozzo eutiroideo
Giudizio fenotipico
Otologico
Le manifestazioni otologiche della BOR sono i sintomi più comuni, con oltre il 90% delle persone in almeno uno dei seguenti:
1. La sordità può presentarsi come trasmissiva, neurosensoriale o mista. La gravità della sordità è variabile, che vanno dalla perdita dell'udito da lieve a profonda. Inoltre, l'entità della perdita dell'udito può essere stabile o progressiva. La perdita dell'udito è ormai consolidata come il tratto singolo più comune tra quelli con la sindrome BOR, stimata in 89% degli individui in uno studio di Fraser et al. Tra quelli con perdita dell'udito, il 50% presentava ipoacusia mista, 30% trasmissiva de il 20% neurosensoriale. E 'stato inoltre stimato che il 2% dei bambini con sordità profonda hanno BOR.
2. Deformità del padiglione .
3. Bozzi preauricolari.
4. Appendici preauricolari.
5 Stenosi del condotto uditivo esterno:
6. Malformazioni degli ossicini dell'orecchio medio, quali ipoplasia o spostamento
7 Orecchio interno: coclea ipoplasica, canali semicircolari laterali displasici, l'allargamento degli acquedotti vestibolari o cocleari.
Anomalie Arco Branchiale
1. Cisti branchiali leporine.
2. Brancial tratto del seno leporino.
3. Branchiale fistole leporino.
Malformazioni renali
1. Uno spettro di malformazioni renali è possibile, da ipoplasia al agenesia.
2.Cisti caliceale.
3. Uretero-pelvica ostruzione del giunto.
4. Reflusso vescico-ureterale
Altri risultati associati
1. Prolasso della valvola mitrale
2. Palatoschisi
3. Aplasia del dotto lacrimale
4. Paralisi del nervo facciale
5. Retrognazia
Studi Gene
Attualmente, vi sono tre mutazioni genetiche conosciuti che provocano sindrome BOR. Se viene effettuata una diagnosi clinica di sindrome di BOR, la conferma dovrebbe essere tentato attraverso l'analisi di sequenza per EYA1. Se una mutazione EYA1 non viene trovato con l'analisi di sequenza, si dovrebbe utilizzare l'analisi duplicazione / cancellazione per EYA1 e analisi di sequenza di SIX 1 e SIX 5.
Mutazione EYA1
· Circa il 40% delle persone con sindrome di BOR avrà una mutazione EYA1.
· EYA1 codifica per un cofattore trascrizione che si esprime nel mesenchima metanefrico durante lo sviluppo dei reni.
SIX1 mutazione
· La mutazione SIX1 è stimata essere trovato 2% dei casi di sindrome BOR.
· Codifica fattori di trascrizione che controllano l'espressione di GDNF e PAX2.
· La maggior parte dei pazienti con questa mutazione non dimostrano malformazione renale.
SIX5 mutazione
· La mutazione SIX5 è presente in circa il 2,5% dei casi di sindrome di BOR.
· Udienza può essere normale in questi pazienti.
Gestione
Valutazione Otologic
· Le prove devono comprendere ABR, audiometria tonale, e la prova di emissione.
· Imaging delle ossa temporali con una TAC è giustificata.
· Una valutazione annuale uditivo dovrebbe essere suggerito.
Valutazione dell’Arco Branchiale
· L'esame fisico seguita dalla tomografia computerizzata (TC), fistologramma, o risonanza magnetica (MRI) se le masse sono palpate .
Valutazione renale
· Ecografia renale
· BUN e creatinina
· I pazienti devono seguire un follow-up annualmente con la nefrologia e urologia.
La consulenza genetica
· È importante che i pazienti affetti da BOR tengano conto conto che c'è una probabilità del 50% di trasmissione per ogni bambino a causa autosomica dominante.
· Test prenatale per BOR è disponibile per quelli con una storia familiare positiva.
Trattamento
Anomalie otologici
· Rilevare eventuale ipoacusia, utilizzare il trattamento acustico appropriato come indicato dalla gravità della perdita, come la protesi o l'impianto cocleare dell'udito.
· Se l'orecchio medio è intatto, con stenosi del canale uditivo esterno, si può prendere in considerazione una canaloplastica.
· Considerate le procedure di chirurgia plastica per la deformità della Pinna.
Anomalie branchiali
· Asportazione di cisti, fistole, o del seno, con dissezione per tutta la lunghezza del tratto e la tonsillectomia.
Anomalie renali
· Il trattamento dipende dalla gravità delle complicanze renali, ma sia la gestione chirurgica e medica dovrebbe essere utilizzato.
· Il trapianto renale può essere considerato se la malattia renale allo stadio terminale si sviluppa.
Manifestazioni generali
La displasia renale è riportato in oltre due terzi dei pazienti e varia da pali rastremati superiori (duplicazione del sistema di raccolta) alla marcata agenesia del rene. La sindrome può includere sequenza Potter.
Dove posso trovare ulteriori informazioni sulla sindrome branchiootorenale?
Si possono trovare le seguenti risorse sulla sindrome branchiootorenal utili. Questi materiali sono scritti per il pubblico in generale.
• MedlinePlus - Informazioni sanitarie (4 collegamenti)
• Malattie Genetiche e Rare Information Center - Informazioni sulle malattie genetiche e le malattie rare
• Ulteriori NIH Risorse - National Institutes of Health
National Institute on Deafness e altri disturbi della comunicazione (NIDCD)
• Risorse educative - Pagine di informazioni (5 collegamenti)
• Supporto paziente - Per i pazienti e le famiglie (3 collegamenti)
Potreste essere interessati a queste risorse, che sono progettati per gli operatori sanitari e ricercatori.
• Gene Recensioni - Sintesi clinica
• Test genetici Registry - archivio di informazioni test genetico (2 collegamenti)
• PubMed - La letteratura recente
• OMIM - Catalogo Malattia genetica (2 collegamenti)
Quali altri nomi la gente usa per la sindrome branchiootorenale?
• BOR
• Sindrome BOR
• Displasia Branchiootorenale
• Branchio-Otorenale Displasia
• Branchio-Otorenale Syndrome
• Sindrome Branchio-Oto-Renale
• Sindrome di Melnick-Fraser
Per ulteriori informazioni sulla denominazione condizioni genetiche, vedere la Genetics Home Reference Condizione Linee guida di denominazione e di come sono condizioni genetiche e geni denominati? nel manuale.
Cosa succede se ho ancora domande specifiche sulla sindrome branchiootorenal?
Chiedi alla Genetica e Malattie Rare Information Center .
References/Suggested Reading
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M,
Amer I, Falzon A, Choudhury N, Ghufoor K. Branchiootic syndrome-a clinic case report and review of the literature. J Pediatr Surg . 2012;47:1604-1606.
Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet1997;15:157–64.[CrossRef][Medline][Web of Science]
Blanck C, Kohlhase J, Engels S, Burfeind P, Engel W, Bottani A, Patel MS, Kroes HY, Cobben JM. Three novel SALL1 mutations extend the mutational spectrum in Townes-Brocks syndrome: clinical and genetic implications. Am J Hum Genet2000;66:1715–20.[CrossRef][Medline][Web of Science]
Borsani G, DeGrandi A, Ballabio A, Bulfone A, Bernard L, Banfi S, Gattuso C, Mariani M, Dixon M, Donnai D, Metcalfe K, Winter R, Robertson M, Axton R, Brown A, Van Heyningen V, Hanson I. EYA4, a novel vertebrate gene related to Drosophila eyes absent. Hum Mol Genet1999;8:11–23.[Abstract/FREE Full text]
Buck A, Kispert A, Kohlhase J. Embryonic expression of the murine homologue of SALL1, the gene mutated in Townes-Brocks syndrome. Mech Dev2001;104:143–6.[CrossRef][Medline][Web of Science]
Chen A, Francis M, Ni L, Cremers C, Kimberling W, Sato Y, Phelps P, Bellman S, Wagner M, Pembrey M, Smith RJ. Phenotypic manifestations of branchiootorenal syndrome. Am J Med Genet . 1995;58:365-370.
Chang
EH, Menezes M, Meyer NC, Cucci RA, Vervoot VS, Schwartz CE, Smith RJ.
Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic
consequences. Hum Mutat . 2004;23:582-9. PubMed
citation ![]()
Correa-Cerro LS, Kennerknecht I, Just W, Vogel W, Müller D. The gene for branchio-oculo-facial syndrome does not colocalize to the EYA1-4 genes. J Med Genet2000;37:620–3.[FREE Full text]
Engels S, Kohlhase J, McGaughran J. A SALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet2000;37:458–60.[FREE Full text]
Fraser FC, Sproule JR, Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet . 1980;7:341-349.
Fujimoto A, Lipson M, Lacro RV, Shinno NW, Boelter WD, Jones KL, Wilson MG. New autosomal dominant branchio-oculo-facial syndrome. Am J Med Genet1987;27:943–51.[CrossRef][Medline][Web of Science]
Gene
Review: Branchiootorenal Spectrum Disorders This link leads to a site outside
Genetics Home Reference. Kochhar
A, Fischer SM, Kimberling WJ, et al. Branchio-oto-renal syndrome. Am J Med
Genet. 2007;15(143A(14)):1671-8, Review. PubMed
citation ![]()
Kumar S, Kimberling WJ, Weston MD, Schaefer BG, Berg MA, Marres HA, Cremers CW. Identification of three novel mutations in human EYA1 protein associated with branchio-oto-renal syndrome. Hum Mutat1998;11:443–9.[CrossRef][Medline][Web of Science]
Kumar S, Deffenbacher K, Marres HAM, Cremers CWRJ, Kimberling WJ. Genomewide search and genetic localization of a second gene associated with autosomal dominant branchio-oto-renal J Hum Genet2000;67(suppl 2):401.
Legius E, Fryns JP, Van den Berghe H. Dominant branchial cleft syndrome with characteristics of both branchio-oto-renal and branchio-oculo-facial syndrome. Clin Genet1990;37:347–50.[Medline]
Kumar S, Marres HAM, Cremers CWRJ, Kimberling WJ. Mutation analysis of EYA1 gene associated with
branchio-oto-renal syndrome and refining the BOR2 region on chromosome 1q. Am syndrome. J Med Genet2000;37:303–7.[FREE Full text]
Lin AE, Doherty R, Lea D. Branchio-oculo-facial and branchio-oto-renal syndromes are distinct entities. Clin Genet1992;41:221–3.[Medline]
Lin AE, Gorlin RJ, Lurie IW, Brunner HG, Van der Burgt I, Naumchik IV, Rumyantseva NV, Stengel Rutkowski S, Rosenbaum K, Meinecke P, Müller D. Further delineation of the branchio-oculo-facial syndrome. Am J Med Genet1995;56:42–59.[CrossRef][Medline][Web of Science]
Lin AE, Semina EV, Daack-Hirsch S, Roeder ER, Curry CJ, Rosenbaum K, Weaver DD, Murray JC. Exclusion of the branchio-oto-renal syndrome locus (EYA1) from patients with branchio-oculo-facial syndrome. Am J Med Genet2000;91:387–90.[CrossRef][Medline]
McKusick VA. Mendelian inheritance in man. Catalogs of human genes and genetic disorders. 12th ed. Baltimore: Johns Hopkins University Press, 1998.
Mishima N, Tomarev S. Chicken eyes absent 2 gene: isolation and expression pattern during development. Int J Dev Biol1998;42:1109–15.[Medline][Web of Science]
Ruf RG, Xu PX, Silvius D, Otto EA,
Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM Jr, Brophy
PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ,
Stratakis CA, Weil D, Petit C, Hildebrandt F. SIX1 mutations cause branchio-oto-renal
syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci US A. 2004 May
25;101(21):8090-5. Epub 2004 May 12. PubMed
citation ![]()
Sahly I, Andermann P, Petit C. The zebrafish eya1 gene and its expression pattern during embryogenesis. Dev Genes Evol1999;209:399–410.[CrossRef][Medline][Web of Science]
Sambrook J, Fritsch EF, Maniatis
T. Molecular cloning. A laboratory manual. 2nd ed. Cold Spring
Harbor, NY: Cold Spring Harbor Laboratory Press, 1989. Sanggaard KM, Rendtorff ND, Kjaer KW,
Eiberg H, Johnsen T, Gimsing S, Dyrmose J, Nielsen KO, Lage K, Tranebjaerg L.
Branchio-oto-renal syndrome: detection of EYA1 and SIX1 mutations in five out
of six Danish families by combining linkage, MLPA and sequencing analyses. Eur J Hum Genet.2007 Nov;15(11):1121-31. Epub 2007 Jul 18. PubMed citation ![]()
Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet1996;58:1323–37.[Medline][Web of Science]
Vincent C, Kalatzis V, Abdelhak S, Chaib H, Compain S, Helias J, Vaneecloo FM, Petit C. BOR and BO syndromes are allelic defects of EYA1. Eur J Hum Genet1997;5:242–6.[Medline][Web of Science]
Xu PX, Cheng J, Epstein JA, Maas RL. Mouse Eya genes are expressed during limb tendon development and encode a transcriptional activation function. Proc Natl Acad Sci USA1997;94:11974–79.[Abstract/FREE Full text]
Wang Y, Treat K, Schroer RJ O'Brien JE, Stevenson RE, Schwartz CE (giugno 1994). "Localizzazione di (BOR) sindrome Branchio-oto-renale a una regione del cromosoma 3 Mb 8q". Am. J. Med.. Genet. 51 (2):. 169-75
Waardenburg sindrome (anche sindrome di Shah Waardenburg, sindrome di Waardenburg-Klein, sindrome di Mende II, Van der sindrome Hoeve-Halbertsma-Waardenburg, sindrome di ptosi-epicanto, Van der sindrome Hoeve-Halbertsma-Gualdi, tipo Waardenburg Pierpont, [5] Van der Hoeve sindrome Waardenburg-Klein, sindrome di Waardenburg II e sindrome di Vogt) è una rara malattia genetica più spesso caratterizzato da diversi gradi di sordità , difetti minori nelle strutture derivanti dalla cresta neurale , e anomalie della pigmentazione. Disagi Nella miogenesi , in particolare mutazioni in Pax3, possono causare la sindrome di Waardenburg I e III.E 'stata descritta per la prima nel 1951.

 Che cosa
è la sindrome di Waardenburg?,pag.2
Che cosa
è la sindrome di Waardenburg?,pag.2
Le cause della sindrome di Waardenburg, pag.3
Fisiopatologia, pag.4
Epidemiologia, pag.4
Chi è a rischio?,pag.4
Trattamento dei sintomi, pag.5
Diagnosi e valutazione audiologica, pag.5
Caratteristiche fisiche,pag.8
È WS, o qualcosa d'altro?, pag.15
Tipi di WS I II II IV, pag.18/23
Malattia di Hirshsprung,pag.23
Che cosa è la sindrome di Waardenburg?
Waardenburg sindrome è una malattia rara caratterizzata da sordità in associazione con anomalie della pigmentazione e difetti dei tessuti cresta di derivazione neurale.
Sindrome è una parola che descrive un gruppo di caratteristiche fisiche che si verificano insieme. Le persone che hanno WS di solito hanno un aspetto unico. Questa unicità è causato da cambiamenti nel loro corredo genetico.
Sindrome di Waardenburg è una condizione genetica che causa una persona di essere nato con le caratteristiche facciali inusuali e perdita dell'udito. Questi problemi fisici possono comparire nel corso della vita della persona. Non tutti con WS avrà tutte le caratteristiche della sindrome, e non tutti avranno una perdita uditiva, ma vi è una maggiore probabilità che lo faranno.
Hammerschlag, nel 1907, e Urbantschitsch, nel 1910, entrambi citano eterocromia dell’iride, albinismo parziale con complicanze di sordomutismo Nel 1916, van der Hoeve descrive una distopia angoli palpebrali mediale lateroversa in una coppia di gemelle monozigoti con Sordomutismo La Sindrome di Waardenburg è stato identificato nel 1951 da Petrus Johannes Waardenburg(1886-1979), un oculista olandese che ha notato che molti dei suoi pazienti avevano caratteristiche facciali uniche, occhi di colore diverso avevano pure, spesso, la perdita dell'udito. In effetti, una caratteristica primaria di Waardenburg è perdita di udito - insieme con gli occhi blu/ marroni o due brillanti occhi azzurri. Un'altra caratteristica facilmente riconoscibile è capelli bianchi sulla testa. Da allora, sono stati descritti quattro tipi specifici di WS. La condizione ha descritto è ora classificato come WS1.
ha definito la sindrome con le seguenti 6 principali caratteristiche
- Spostamento laterale della angoli palpebrali mediali combinata con distopia del punto lacrimale e blefarofimosi
- Aspetto di Eyes Wide-set a causa di una prominente, larga radice del naso(distopia dei canti ),associata in particolare con il tipo I),nota anche come telecanto ;
- Ipertricosi della parte mediale delle sopracciglia che toccano nel mezzo
- Un ciuffo di capelli bianchi ( poliosi ), o ingrigimento precoce dei capelli;
- Occhi molto chiari o azzurri brillanti, occhi di due colori diversi (eterocromia completa ), o gli occhi con un’iride con due colori diversi (eterocromia settoriale);
- Sordità da moderata a profonda (frequenza più alta associata al tipo II);
L’oculista svizzero David Klein, nel 1947, ha anche fatto contributi per la comprensione della sindrome,Klein ha riportato il caso di una ragazza di 10 anni con deafmutism, albinismo parziale della pelle e dei capelli, iridis ipocromia, blefarofimosi con ipertelorismo e l'assenza dell'angolo nasofrontal, ipertricosi delle sopracciglia, e molteplici anomalie associate (mio -osteo-Articulare displasia).
WS2 è stato identificato nel 1971, per descrivere i casi di " distopia dei canti "non era presente. [4] WS2 è ora diviso in sottotipi, basato sul gene responsabile.
Sono stati identificati altri tipi, ma sono meno comuni.
Mutazioni nel EDN3, EDNRB, MITF, PAX3, SNAI2, e SOX10 sono i geni che sono affetti dalla sindrome di Waardenburg. Alcuni di questi geni sono coinvolti nella realizzazione dei melanociti, che rende il pigmento melanina. La melanina è un pigmento importante nello sviluppo di capelli, il colore degli occhi, della pelle, e le funzioni dell'orecchio interno. Così la mutazione di questi geni può portare a pigmentazione anomala e la perdita dell'udito. [5] PAX3 e MTIF mutazione del gene avviene in tipo I e II (WS1 e WS2). Tipo III (WS3) mostra mutazioni del gene PAX3 anche. Mutazioni del gene SOX10, EDN3, o EDNRB si verificano in tipo IV.Tipo IV (WS4) può anche interessare porzioni di sviluppo delle cellule nervose che potenzialmente possono portare a problemi intestinali.
Le cause della sindrome di Waardenburg
Questa condizione è solitamente ereditata come autosomica dominante modello, il che significa una copia del gene alterato è sufficiente a causare il disturbo. Nella maggior parte dei casi, una persona affetta ha un genitore con la condizione. Una piccola percentuale di casi derivano da nuove mutazioni nel gene; questi casi si verificano in persone senza storia di malattia nella loro famiglia.
Alcuni casi di tipo II e sindrome tipo IV Waardenburg sembrano avere un autosomica recessiva modello di ereditarietà, il che significa due copie del gene devono essere modificati per una persona di essere colpiti dalla malattia. Molto spesso, i genitori di un bambino con una malattia autosomica recessiva non sono interessati, ma sono portatori di una sola copia del gene alterato.
·

Waardenburg sindrome è solitamente ereditata come modello autosomico dominante .
·

Tipi II e sindrome IV Waardenburg possono talvolta avere un modello di ereditarietà autosomico recessivo .
I geni sono responsabili per dirigere la crescita e lo sviluppo degli esseri umani. Quando i geni sono mutati, o diverso dal normale, possono causare cambiamenti nella normale crescita e sviluppo. In WS, i geni mutati sono responsabili di distribuzione del pigmento nel corpo di sviluppo embrionale e fetale. Questi geni possono causare pigmento da distribuire in modo anomalo. Quando questo accade, alcune zone del corpo possono avere troppo o troppo poco pigmento, o nessun pigmento affatto. Alterazioni della pigmentazione possono produrre occhio inusuale, la pelle, o il colore dei capelli così come la sordità. Gli scienziati non hanno ancora capito esattamente che cosa causa le mutazioni genetiche che producono WS, anche se sono stati identificati i geni stessi.
Fisiopatologia
La sindrome Waardenburg è una malattia rara con una modalità autosomica dominante. Diverse ipotesi sono state avanzate per spiegare tutte le caratteristiche cliniche della sindrome.
- La teoria della cresta neurale carente, suggerisce una anomalia dello sviluppo della cresta neurale come causa di malattia: L'associazione della sindrome di Waardenburg e megacolon congenito aganglionico supporta questa ipotesi.
- La sindrome di Waardenburg come una parte della sindrome del primo arco
- Un rapporto della sindrome di Waardenburg con lo status dysraphicus
- La teoria della necrosi intrauterina
Nessuna di queste possibilità spiega tutte le caratteristiche della sindrome di Waardenburg. Cause ereditarie rappresentano circa il 50% degli individui osservati per l'infanzia (prelinguale) perdita di udito, di cui il 70% sono dovuti a mutazioni in numerosi geni singoli che compromettono la funzione uditiva solo (non sindromica). Il resto sono associate ad altre anomalie dello sviluppo chiamati sordità sindromica.
I Geni responsabili di forme sindromiche di perdita dell'udito nella sindrome di Waardenburg includono PAX3 sulla banda 2q37, osservata nei tipi I e III, e MITFmappato su 3p12-p 14,1 per il tipo II(Tachibana M.,1997; Zhang H et al., 2012; Wildhardt G et al 2013 . La sindrome di Waardenburg è autosomica dominante per la maggior parte delle persone con tipo I, II, e III. La sindrome di Waardenburg di tipo IV è autosomica recessiva con penetranza variabile ed è causa di mutazioni del gene SOX10 o del recettore dell'endotelina-B ( EDNRB ), che sembrano correlare con l'intestinale e / o sintomi neurologici manifesta nei pazienti(Morell R, et al.1998;DeStefano AL, et al.,1998;Bondurand N, et al.,2000; Dundar M, et al.,2001;Chan KK, et al.,2003; Sznajer et al.,2008; Oshimo T, et al.,2012;Jung HJ, et al.,2013;hanno descritto un sito di splice mutazione SOX10 (c.698-2A> C), che ha provocato una sindrome di Waardenburg grave di tipo 4 senza malattia di Hirschsprung . Il bambino presentava vivaci occhi blu, ritardo mentale, sinofria, sordità, canali completi bilaterali semicircolari, e neuropatia periferica.
Epidemiologia
Quanto rara è la sindrome di Waardenburg?
La frequenza della sindrome Waardenburg è stimato a 1 caso ogni 212.000 persone nella popolazione generale dei Paesi Bassi, ma a causa di una bassa penetranza di circa il 20%, la frequenza dell'intero sindrome (con o senza sordità) è probabilmente di circa 1 caso per ogni 42.000 persone. In Kenya, Secondo il National Institutes of Health, un individuo su 10.000 a 20.000 avrà la sindrome di Waardenburg. Inoltre, dal momento che i primi due tipi (tipo I e II) sono più comuni e sono associati con la perdita dell'udito, si può presumere che la maggior parte degli individui con Waardenburg avrà la perdita dell'udito.
La sindrome è stata osservata in 0,9-2,8% delle persone con sordomutismo.
Mortalità / morbilità
I bambini con sindrome di Waardenburg hanno una aspettativa di vita normale. La morbilità è legato alla sordità e difetti dei tessuti cresta di derivazione neurale, tra cui ritardo mentale, convulsioni, disturbi psichiatrici, anomalie scheletriche, e disturbi oculari (cataratta comprese).
Razze
Waardenburg sindrome colpisce persone di tutte le razze di tutto il mondo.
Sesso
La malattia colpisce ugualmente entrambi i sessi. Non sono stati trovati differenze sessuali tra persone con sordomutismo congenito.
Età
Come una malattia ereditaria, la sindrome di Waardenburg può essere riconosciuto immediatamente o subito dopo la nascita. Alcune caratteristiche dermatologiche (ad esempio, poliosi) cambiano con l'età.
Si stima che circa 1 su 4000 individui nascono con WS. WS si verifica in tutte le razze, e si verifica in modo uguale in maschi e femmine. Nella maggior parte dei tipi di WS, c'è una probabilità del 50% che un bambino nato da una madre con un gene normale e un genitore con il gene WS avrà WS. Chiunque può nascere con WS come risultato di una nuova mutazione genetica.
Il trattamento dipende dal fenotipo dell'individuo con Sindrome di Waardenburg. È possibile ottenere un trattamento per uno dei seguenti segni fenotipiche:
1) Sordità da moderata a profonda (frequenza più alta associata al tipo II);
2) La malattia di Hirschsprung (HD)
3) problemi estetici
4) palatoschisi
5) Mano e contratture delle dita
Il trattamento per queste caratteristiche comporta la consulenza genetica e / o rinvio di appropriarsi medici professionisti come un audiologo, logopedista, fisioterapista e chirurgo plastico.
Diagnosis and Audiological Evaluation Diagnosi e valutazione audiologica
Educational Opportunities Opportunità educative
Americans with Disabilities Act (ADA) Americans with Disabilities Act (ADA)
Literature and Multimedia Letteratura e Multimedia
Diagnosi
La perdita dell'udito può essere trattata, l'udito deve essere controllato da un audiologo. Lui o lei determinerà il grado e il tipo di perdita uditiva. Ecco un elenco di alcuni dei test che un audiologo può somministrare. Non tutti hanno bisogno di tutti questi test. Il medico o l'audiologo può raccomandare quelli necessari, in base alla storia familiare ei sintomi della perdita dell'udito.
Valutazione audiologica
1. Otoscopia
 L’Otoscopia è un
esame fisico del vostro orecchio esterno, condotto uditivo, e timpano. Un audiologo userà un otoscopio per cercare
eventuali perforazioni del timpano, arrossamento del timpano, cerume o segni di
infezione.
L’Otoscopia è un
esame fisico del vostro orecchio esterno, condotto uditivo, e timpano. Un audiologo userà un otoscopio per cercare
eventuali perforazioni del timpano, arrossamento del timpano, cerume o segni di
infezione.
2. Timpanometria
 La timpanometria è una misura della rigidità del
timpano e ci dice come gli ossicini dell'orecchio medio funzionano. Questo test consente di rilevare liquido
nell'orecchio medio, rottura o eventuali lussazioni degli ossicini ,che si
trovano nell'orecchio medio, un perforazione nel timpano, e una malattia
dell'orecchio medio chiamato otosclerosi. L'audiologo metterà una sonda morbida nel
canale uditivo e una macchina rilascia una piccola quantità di pressione. Il
movimento strumento misura le risposte del timpano alle variazioni di pressione
e registra il risultato su un grafico.
La timpanometria è una misura della rigidità del
timpano e ci dice come gli ossicini dell'orecchio medio funzionano. Questo test consente di rilevare liquido
nell'orecchio medio, rottura o eventuali lussazioni degli ossicini ,che si
trovano nell'orecchio medio, un perforazione nel timpano, e una malattia
dell'orecchio medio chiamato otosclerosi. L'audiologo metterà una sonda morbida nel
canale uditivo e una macchina rilascia una piccola quantità di pressione. Il
movimento strumento misura le risposte del timpano alle variazioni di pressione
e registra il risultato su un grafico.
3. Audiometria Tonale ai Toni Puri
 Il test ai toni puri è
il tipo più comune di valutazione dell’udito.
L'audiologo
può utilizzare cuffie o inserire gli auricolari, che sono come tappi per le
orecchie, per la valutazione della conduzione per via aerea. Durante il test, si sentono toni a diverse
frequenza ed intensità, e l’esaminato deve alzare la mano quando sente il
suono.
L'audiologo
registra le soglie per ogni frequenza su un grafico chiamato audiogramma. La soglia è il livello
più basso di intensità a cui si possono sentire i toni puri. Il test ai toni puri
comprende anche il test di conduzione per via ossea.
Durante
questo tipo di test, si indossa una fascia sulla testa con vibratore appoggiato
sulla mastoide , mentre sono presentati i toni. È possibile ascoltare i suoni che vengono
condotti all’orecchio interno attraverso il cranio. Un audiologo poi confrontare i risultati
dei toni puri condotti per via aerea ,con quelli trasmessi per via ossea e
determinare quale parte dell’orecchio è responsabile di qualsiasi perdita
uditiva.
Il test ai toni puri è
il tipo più comune di valutazione dell’udito.
L'audiologo
può utilizzare cuffie o inserire gli auricolari, che sono come tappi per le
orecchie, per la valutazione della conduzione per via aerea. Durante il test, si sentono toni a diverse
frequenza ed intensità, e l’esaminato deve alzare la mano quando sente il
suono.
L'audiologo
registra le soglie per ogni frequenza su un grafico chiamato audiogramma. La soglia è il livello
più basso di intensità a cui si possono sentire i toni puri. Il test ai toni puri
comprende anche il test di conduzione per via ossea.
Durante
questo tipo di test, si indossa una fascia sulla testa con vibratore appoggiato
sulla mastoide , mentre sono presentati i toni. È possibile ascoltare i suoni che vengono
condotti all’orecchio interno attraverso il cranio. Un audiologo poi confrontare i risultati
dei toni puri condotti per via aerea ,con quelli trasmessi per via ossea e
determinare quale parte dell’orecchio è responsabile di qualsiasi perdita
uditiva.
4. Emissioni Otoacoustiche (OAE)
La coclea è la parte dell'orecchio che riceve informazioni dai suoni in entrata e invia i suoni al cervello. La coclea contiene molte terminazioni nervose chiamate "cellule ciliate". Quando questi terminazioni nervose lavorano correttamente, i suoni vengono inviati al cervello in maniera accurata ed il cervello interpreta questi suoni come la musica, la parola, o il rumore. Le coclee normali hanno le cellule ciliate esterne che producono rumore. Il test’s OAE registra i suoni che l'orecchio produce e aiuta l'audiologo a determinare se il vostro orecchio interno trasmette il suono al cervello.
5. Auditory Brainstem Response (ABR)
Il test ABR è uno strumento diagnostico non invasivo che misura le onde cerebrali in risposta ad uno stimolo uditivo. Esso misura l'attività della via uditiva che si trova nel vostro tronco encefaloo Per ulteriori informazioni sui test ABR, andare a questi siti.:
www.med.umich.edu www.med.umich.edu
www.hearingcenter.com www.hearingcenter.com
Le prove di cui sopra sono solo alcuni dei possibili modi in cui gli audiologi determinano se l'udito è normale. Per saperne di più sulla diagnosi della perdita dell'udito, visitare i seguenti siti web:
American Speech and Hearing Association American Speech and Hearing Association
Children's Hopsital Boston Bambini Hopsital Boston
Le opzioni per il trattamento WS variano a seconda delle particolari caratteristiche WS. Parte di udito trattamento di perdita è l'intervento di un consulente genetico. Il trattamento può includere uno o una combinazione delle seguenti opzioni. Ricordate che il trattamento (s) scelto da una persona con WS, non può essere adatto per voi o il vostro bambino.
Genetic Testing & Counseling 1. Genetic Testing & Counseling
2. Opzioni per il trattamento di perdita di udito includono:
- Apparecchi acustici o Amplification
- Impianti cocleari
- Sign Language
- Logoterapia
Voi o il vostro bambino può scegliere tra una varietà di scuole per sentire gli individui con problemi. Considerate le esigenze specifiche del vostro bambino e il grado di perdita di udito quando si sceglie una scuola. Molte scuole specializzate in formazione per le persone con problemi di udito. Alcune scuole si affidano completamente sulla lingua dei segni americana, alcuni includono una combinazione del linguaggio parlato e segni orale, e altri usano il discorso solo orale. Oltre alle scuole di specializzazione, si può scegliere di inviare il vostro bambino a una scuola pubblica all'interno della vostra comunità. Individuals with Disabilities Education Act (IDEA) 'Gli Stati Uniti individui con disabilità Education Act (IDEA) afferma che "ogni bambino ha diritto a una istruzione gratuita e adeguata nel contesto meno restrittivo." In altre parole, il bambino deve essere dotato delle risorse adeguate in una scuola vicino te.
Caratteristiche fisiche
Caratteristiche fisiche dei WS sono classificati come maggiore o minore. Per essere diagnosticati con WS una persona deve dimostrare due caratteristiche principali, o una maggiore e due minori caratteristiche della sindrome. Non tutti coloro che hanno WS avrà tutte queste caratteristiche, e non tutte le caratteristiche della sindrome sarà presente alla nascita. Alcune caratteristiche si sviluppano più tardi nella vita, e alcuni diventano più pronunciati con l'età.
Le principali caratteristiche sono elencate di seguito. Le descrizioni di ciascuna si trovano qui .
• Eterocromia Iridis
• luminosi occhi azzurri
• Distopia dei canti (spostamento laterale angoli interni palpebrali )
• Broad, radice del naso prominente
• Piccolo mid-face
• capelli prematuramente grigi
• sordità neurosensoriale congenita
• WS Tipo 4 & Malattia di Hirschsprung
Eterocromia Iridis
|
|
|

Occhi molto chiari o brillanti azzurri, occhi di due colori diversi (eterocromia completa), o gli occhi con un’ iride con due colori diversi (eterocromia settoriale);"Eterocromia Iridis" significa che qualcuno, o un animale, ha gli occhi di due colori diversi, come fa il gatto nella foto a destra. Può anche significare due colori nella stessa dell'occhio, come nella foto a sinistra. Questa persona è nata con gli occhi azzurri, e sviluppato macchie di marrone durante l'adolescenza.
Eterocromia rende occhi sguardo insolito, ma non influenza la visione della persona. WS solito non causare problemi di visione.
È interessante notare che gli animali, tra cui cavalli, cani, topi e gatti possono avere WS. Animali sordi fanno i buoni animali domestici, ma sono spesso distrutti nella convinzione che essi non possono essere addestrati. Animali sordi possono fare buoni animali domestici, se sono tenuti in un ambiente protetto.
|
|
|


Le persone con WS hanno spesso occhi blu brillanti belli, in combinazione con una o più delle altre caratteristiche di WS. Occhi azzurri e prematuramente gray hair often occur in combination, as they do in the photo to the left. capelli grigi si verificano spesso in combinazione, come fanno nella foto a sinistra. Qualsiasi persona con gli occhi azzurri può avvertire fotofobia, difficoltà di vedere alla luce del sole, perché le persone con gli occhi azzurri non hanno lo strato sovrastante di pigmento scuro che le persone con marrone, nocciola, o gli occhi verdi fanno. Questo pigmento aggiuntivo protegge gli occhi dalla luce forte. Le persone con gli occhi azzurri anche spesso hanno scarsa visione notturna. Gli occhi azzurri da soli non sono diagnostici di WS; tuttavia, gli occhi azzurri in combinazione con i capelli precocemente grigi, aree di pelle depigmentazione, perdita dell'udito suggeriscono WS.
Back to Top Distopia dei canti
|
|
"Distopia dei canti" descrive l'aspetto degli occhi della persona e ponte nasale. Le persone con distopia dei canti hanno ampie, ponti nasali piatte, con pieghe di pelle che copre angoli interni degli occhi. Queste pieghe della pelle danno un aspetto asiatico agli occhi di persone con WS. Persone asiatici come quello nella foto a destra, hanno pieghe cutanee copre gli angoli interni degli occhi chiamato epicanto. Queste pieghe sono una caratteristica del viso normale causata dai ponti nasali pianeggianti tipici di persone asiatiche. Dystopia dei canti, come mostrato nella foto a fianco, non è una condizione normale per persone di ogni razza. I medici utilizzano una formula denominata "W Index" per calcolare la distanza tra gli occhi per stabilire se qualcuno ha distopia dei canti.
Back to Top Broad Nasal Root
Broad nasale Root
 importanti radici
nasali si trovano spesso nelle persone con WS.
La radice nasale è l'area tra gli occhi del ponte della occhiali poggia. Le persone
con importanti radici nasali sembrano avere piani, facce larghe, ei loro occhi
possono apparire da impostare ampiamente a parte. La persona nella foto ha anche le
brillanti occhi azzurri, volumi bruciati sopracciglia, piccolo alae nasale, e
caratteristici nevi di WS.
importanti radici
nasali si trovano spesso nelle persone con WS.
La radice nasale è l'area tra gli occhi del ponte della occhiali poggia. Le persone
con importanti radici nasali sembrano avere piani, facce larghe, ei loro occhi
possono apparire da impostare ampiamente a parte. La persona nella foto ha anche le
brillanti occhi azzurri, volumi bruciati sopracciglia, piccolo alae nasale, e
caratteristici nevi di WS.
 Le persone con WS
possono avere ossa facciali che sono più piccoli del normale. Queste
ossa sono le ossa nasali, guancia ossa, e alcune ossa che formano le
occhiaie. Quando ossa metà
viso sono più piccolo del previsto, il viso della persona può apparire piatta
nel profilo. Ossa nasali appiattite possono causare gli
occhi della persona a comparire ampiamente impostare e
"orientale-looking", anche se la persona non è asiatico.
Le persone con WS
possono avere ossa facciali che sono più piccoli del normale. Queste
ossa sono le ossa nasali, guancia ossa, e alcune ossa che formano le
occhiaie. Quando ossa metà
viso sono più piccolo del previsto, il viso della persona può apparire piatta
nel profilo. Ossa nasali appiattite possono causare gli
occhi della persona a comparire ampiamente impostare e
"orientale-looking", anche se la persona non è asiatico.
Quest'uomo ha piccole ossa della faccia media, eterocromia dell’iridis e capelli precocemente grigi - tutte le caratteristiche di WS.
Capelli prematuramente grigi, o macchie di capelli grigi nei capelli di colore più scuro sono anche segni di WS. Molte persone con WS hanno una striscia bianca di capelli al centro della loro fronte. Altri possono avere i capelli che si è interamente grigio quando erano adolescenti. Alcune persone con WS hanno ciglia e sopracciglia bianche. I capelli grigi si verifica spesso in combinazione con gli occhi blu e la perdita dell'udito. Persone i cui capelli sono diventati grigi prematuramente possono scegliere di tingersi i capelli, quindi questa funzione non è sempre visibile in qualcuno con WS. L'uomo nella foto (in alto a destra) ha i capelli che sono diventati grigi quando era adolescente.
Sordità neurosensoriale
 hearing
loss
Ipoacusia neurosensoriale
può essere presente alla nascita (congenita), o può apparire più tardi nella
vita. Nei casi più gravi soggetti con WS sono nati sordi. Alcuni individui hanno la perdita
dell'udito in un solo orecchio, e alcuni individui hanno perdite uditive molto
lievi, che non interferiscono con la parola o lo sviluppo del linguaggio. Poiché la perdita dell'udito non è una caratteristica
visibile, le persone che hanno alcune delle altre caratteristiche di WS devono
sempre ricevere una valutazione dell'udito. Un ragazzo con un impianto
cocleare è raffigurato qui sotto.
hearing
loss
Ipoacusia neurosensoriale
può essere presente alla nascita (congenita), o può apparire più tardi nella
vita. Nei casi più gravi soggetti con WS sono nati sordi. Alcuni individui hanno la perdita
dell'udito in un solo orecchio, e alcuni individui hanno perdite uditive molto
lievi, che non interferiscono con la parola o lo sviluppo del linguaggio. Poiché la perdita dell'udito non è una caratteristica
visibile, le persone che hanno alcune delle altre caratteristiche di WS devono
sempre ricevere una valutazione dell'udito. Un ragazzo con un impianto
cocleare è raffigurato qui sotto.
Caratteristiche di minore importanza sono i seguenti, con le descrizioni disponibili qui :
•
leucoderma congenita
• Diverse nevi (moli)
• anomalie sopracciglio
• ipoplasico nasale alae
• Sindattilia
• mento sfuggente
• labio-palatoschisi
• Spina bifida
• Disturbi della comunicazione
• Camptodattilia
• caratteristiche rare
Sindrome di Usher La sindrome Graefe-Usher La sindrome sordità-retinite pigmentosa La sindrome Hallgren
- trasmissione autosomica recessiva
- sordità neurosensoriale congenita (raramente progressiva)
- retinite pigmentosa (RP)
- eterogeneità genetica - Tipi I, Il, III
Che cos'è la sindrome di Usher? Fig. 1
La sindrome di Usher è una condizione genetica che comporta la perdita dell'udito neurosensoriale e retinite pigmentosa (RP). Sebbene sia considerata una malattia rara, è la causa più frequente di sordo-cecità negli esseri umani. Una sindrome è una malattia o un disturbo che ha più di una caratteristica o sintomo. Quando la perdita dell'udito è accompagnata da altri riscontri clinici, è spesso classificato come una sindrome, e ci sono più di 400 tali sindromi descritte da Gorlin, Torell, e Cohen (1995). la sindrome di Usher è una condizione caratterizzata da perdita progressiva dell'udito o sordità e perdita della vista da retinite pigmentosa (RP). La condizione prende il nome da un oculista britannico, CH Usher, che in un articolo nel 1914 descrisse diversi casi in cui si è sottolineato il legame tra sordità congenita e RP. Tuttavia, nel lontano 1860 lavoratori, come von Graef Lo screening uditivo neonatale ha ridotto l'età di identificazione dei bambini con perdita uditiva 12-18 mesi a 6 mesi o meno (Harrison & Rousch, 1996), ma la diagnosi di sindrome di Usher, con la sua devastante perdita della vista, tipicamente ritardi 5-10 anni dietro l'identificazione della perdita dell'udito (Kimberling & Lindenmuth, 2007). Così, mentre i genitori imparano di perdita uditiva del loro bambino relativamente presto, senza diagnosi differenziale si procede con le decisioni critiche relative all'intervento, comunicazione e opzioni educative ignari che il loro bambino finirà per essere cieco. A causa dello screening uditivo neonatale, audiologi sono spesso contatto primario della famiglia. Pertanto, audiologi sono in grado di migliorare i risultati di diagnosi differenziali per i bambini con sindrome di Usher. Questo articolo è rivolto ad accrescere la comprensione audiologi 'della presentazione audiologico e visivo, criteri diagnostici e strategie di intervento coinvolti con la sindrome di Usher.
Storia
La sindrome di Usher è chiamato dopo l'oculista scozzese Charles Usher , che ha esaminato la patologia e la trasmissione di questa malattia nel 1914 sulla base di 69 casi. Tuttavia, è stato descritto nel 1858 da Albrecht von Gräfe , (un pioniere della moderna oftalmologia) e Liebreich a Berlino erano a conoscenza del legame tra sordità congenita e RP, soprattutto nei matrimoni consanguinei.. Egli ha riferito il caso di un paziente sordo con retinite pigmentosa , che aveva due fratelli con gli stessi sintomi. Tre anni più tardi(1861), uno dei suoi studenti, Richard Liebreich , ha esaminato la popolazione di Berlino che avevano sordità con retinite pigmentosa. Liebreich ha notato che la sindrome di Usher era di tipo recessivo, dal momento che i casi di combinazioni cieco sordità si sono verificati in particolare nei fratelli dei matrimoni consanguinei o in famiglie con pazienti in diverse generazioni. Le sue osservazioni hanno fornito le prime prove per la trasmissione accoppiata di cecità e sordità, dal momento che non potrebbe essere trovati casi isolati delle due patologie negli alberi genealogici.
La perdita della vista è causata da una malattia dell'occhio chiamata retinite pigmentosa (RP), che colpisce lo strato di tessuto sensibile alla luce nella parte posteriore dell'occhio (retina). Perdita della vista si verifica in quanto le cellule della retina che rilevano la luce si deteriorano gradualmente. Di solito, i bastoncelli della retina sono i primi ad essere interessati, con conseguente precoce cecità notturna e la graduale perdita di visione periferica. In altri casi, si verifica, la degenerazione precoce dei coni della macula, portando ad una perdita di acuità centrale. In alcuni casi, è risparmiata la visione foveale, portando ad una "visione a ciambella"; visione centrale e periferica sono intatti, ma un anello esiste intorno alla regione centrale in cui visione è compromessa . Inizia prima la perdita della visione notturna, cecità notturna, questo disturbo può anche essere accoppiato alla difficoltà ad adattarsi alla luce intensa od a rapidi cambiamenti delle condizioni di luce, seguita da punti ciechi che si sviluppano nella visione laterale (periferica). Nel corso del tempo, questi punti ciechi si ingrandiscono e si fondono per produrre una visione a tunnel. In alcuni casi di sindrome di Usher, la visione è ulteriormente compromessa dalla opacità del cristallino dell'occhio (cataratta). Molte persone con retinite pigmentosa mantiene una certa visione centrale per tutta la vita, però.
Quanto è diffusa la sindrome di Usher?
Si pensa che la sindrome di Usher possa essere responsabile dal 3 per cento, al 6 per cento di tutta la sordità infantile e circa del 50 per cento dei casi di sordità-cecità negli adulti. Si stima che la sindrome Usher di I tipo colpisce almeno 4 individui ogni 100.000 persone. Negli Stati Uniti, i tipi 1 e 2 sono i tipi più comuni. Insieme, essi rappresentano circa il 90 al 95 per cento di tutti i casi di bambini che hanno la sindrome di Usher. Può essere ancora più comune in alcune popolazioni etniche, come nelle etnie di ascendenza ebraica Ashkenazi (dell'Europa centrale e orientale) e nella popolazione Acadian in Louisiana. Si pensa che la sindrome di Usher di II Tipo sia la forma più comune, anche se la frequenza di questo tipo è sconosciuto. Nella maggior parte delle popolazioni la sindrome di Usher di Tipo III rappresenta solo una piccola percentuale di tutti i casi di sindrome di Usher. Questa forma della malattia ,tuttavia, è più comune nella popolazione finlandese, dove rappresenta circa il 40 per cento di tutti i casi.
I ricercatori hanno individuato tre principali tipi di sindrome di Usher, designati come tipo I, II, e III. Questi tipi si distinguono per la loro gravità e l'età in cui i segni ei sintomi compaiono. Il tipo I è ulteriormente suddivisa in sette distinti sottotipi, indicati come tipi IA/IG. La sindrome di Usher di tipo II ha almeno tre sottotipi descritti, designati come tipi IIA, IIB e IIC.
|
|
|
Fig. 1:Fotografia della retina di un paziente con la sindrome di Usher (a sinistra) rispetto ad una retina normale (a destra). Il nervo ottico (freccia) sembra molto chiaro, i vasi (stelle) sono molto sottili e non vi è pigmento caratteristico, chiamato spicole ossee (doppie frecce).
|
|
|
|
Le malattie genetiche possono essere causate da un cambiamento (s) in un gene. Ogni individuo ha due copie dello stesso gene. Malattie genetiche vengono ereditate in modi diversi. La sindrome di Usher è una malattia recessiva.Fig.2a/b Mezzi Recessiva:
Un individuo con la sindrome di Usher solito:
Un individuo che ha una cambiata Usher gene sindrome è chiamato un vettore. Quando due portatori dello stesso gene sindrome di Usher hanno un figlio insieme, con ogni nascita c'è un:
|
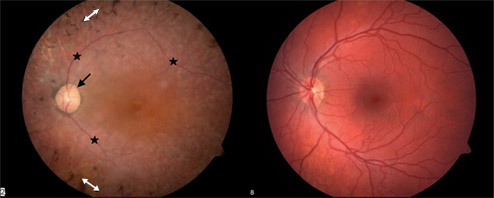
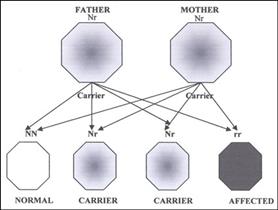
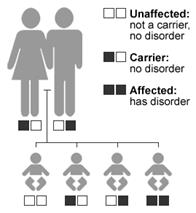
Che cosa provoca la sindrome di Usher?
Sindrome di Usher è ereditata, il che significa che si passa dai genitori ai figli attraverso i geni. Geni sono localizzati in quasi ogni cellula del corpo. I geni contengono le istruzioni che dicono le cellule cosa fare. Ogni persona eredita due copie di ogni gene, uno da ciascun genitore. A volte i geni sono alterati o mutati. Geni mutati possono indurre le cellule a agire in modo diverso rispetto al previsto.
La sindrome di Usher è ereditata come carattere autosomico recessivo. Il termine autosomica significa che il gene mutato non è situato su uno dei cromosomi che determinano il sesso di una persona; in altre parole, maschi e femmine possono avere la malattia e può passarlo insieme ad un bambino. La parola recessiva significa che, per avere la sindrome di Usher, una persona deve ricevere una forma mutata del gene sindrome di Usher da ciascun genitore. Se un bambino ha una mutazione in un gene (sindrome Usher), ma l'altro gene è normale, si prevede che avrà un udito ed una visione normale '. Le persone con una mutazione in un gene che può causare una malattia autosomica recessiva sono chiamati vettori, perché "portano" il gene con una mutazione, ma non mostrano sintomi del disturbo. Se entrambi i genitori sono portatori di un gene mutato per la sindrome di Usher, avranno una probabilità su quattro di avere un figlio con la sindrome di Usher con ogni nascita.
Di solito, i genitori che hanno un udito normale e la visione non sanno se sono portatori di una mutazione del gene (sindrome di Usher). Attualmente, non è possibile determinare se una persona che non ha una storia familiare di sindrome di Usher è un vettore.
Quali sono le caratteristiche dei tre tipi di sindrome di Usher?
Tipo 1
I bambini con diabete di tipo 1, e sindrome di Usher nascono completamente sordi ( Fig. 3a) o perdono gran parte della loro udito entro il primo anno di vita ed hanno gravi problemi di equilibrio. La perdita progressiva della vista ,causata dalla retinite pigmentosa, si manifesta durante l'infanzia. Questo tipo di sindrome di Usher comprende anche problemi dell'orecchio interno che influenzano l'equilibrio. I genitori dovrebbero consultare il proprio medico e altri professionisti della salute dell'udito il più presto possibile per determinare il miglior metodo di comunicazione per il loro bambino. L'intervento dovrebbe essere introdotto presto, durante i primi anni di vita, in modo che il bambino può approfittare della finestra unica di tempo durante il quale il cervello è più ricettivo per l'apprendimento delle lingue, sia parlato o firmato. Se un bambino viene diagnosticato con il tipo 1, sindrome di Usher presto, prima che lui o lei perde la capacità di vedere, quel bambino ha maggiori probabilità di beneficiare di tutta la gamma di strategie di intervento che possono aiutare lui o lei di partecipare più pienamente nelle attività della vita.
La funzionalità vestibolare è caratterizzata da areflessia nella sindrome di Usher Tipo I ,a causa dei problemi di equilibrio associati alla sindrome di Usher di tipo 1, i bambini con questo disturbo di solito associato a areflessia vestibolare e ritardo nelle tappe dello sviluppo, come nel controllo del capo e nell'acquisizione della stazione seduta e della deambulazione autonoma, sono lenti a sedersi senza supporto e di solito non camminano autonomamente prima dei 18 mesi di età(Moller, Kimberling, Davenport, Priluck, & White, 1989). Questi bambini di solito iniziano a sviluppare problemi di visione nella prima infanzia, quasi sempre dal momento in cui raggiungono 10 anni. Questa forma richiede una diagnosi precoce, entro i 2-3 anni, perché un impianto cocleare può ridurre di molto le difficoltà comunicative dovute al sommarsi di difetto uditivo e visivo
Problemi di visione più spesso iniziano con difficoltà a vedere di notte, Cecità notturna è tipicamente appare a 10 anni, quindi questi bambini possono avere paura del buio e sono spesso descritti come goffi perché urtano, o inciampare. Il deterioramento significativo del campo visivo e l'acutezza inizia tra la seconda e la terza decade di vita, con la cataratta essendo una complicanza comune (Edwards, Fishman, Anderson, Boschetto, e Derlackie, 1998; Piazza, Fishman, Sugata, Derlacki, e Anderson, 1986; A. Sadeghi, Eriksson, Kimberling, Sjostrom, e Moller, 2006) ma tendono a progredire rapidamente fino a quando la persona è completamente cieco.
|
|
|
Fig. 2a. Tipici risultati audiometrici nella sindrome di tipo 1 Usher.
|
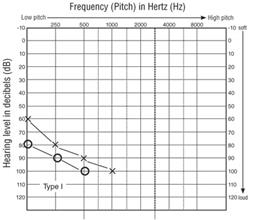
Tipo 2
La sindrome di Usher di tipo 2 (circa il 60% dei casi), è caratterizzata da perdita dell'udito dalla nascita Fig.2b ,(la sordità è meno grave, stabile, prevalentemente sulle frequenze acute) e progressiva perdita della vista che inizia in adolescenza o in età adulta e solitamente il quadro è meno grave rispetto al 1 tipo, non associata a alterazioni vestibolari,. La perdita di udito associata a questa forma di sindrome di Usher varia da una forma lieve ad una grave e colpisce soprattutto i toni alti. I bambini affetti hanno problemi di udito, suoni del linguaggio morbido elevate, come quelle delle lettere S e t. Il grado di perdita dell'udito varia all'interno e tra le famiglie con questa condizione. A differenza di altre forme di sindrome di Usher, le persone con diabete di tipo II non hanno problemi di equilibrio causata da problemi dell'orecchio interno.
|
|
|
Figura 2b. Tipici risultati audiometrici di tipo II sindrome di Usher.
|
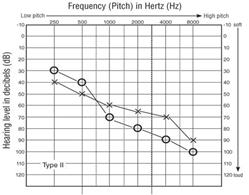
Tipo 3
I bambini con sindrome di Usher tipo 3 (<3% dei casi, diffusa soprattutto nelle popolazioni Finlandesi e negli Ebrei Ashkenaziti), hanno un udito normale alla nascita, che progredisce durante l'adolescenza ,dopo lo sviluppo del linguaggio, richiedono apparecchi acustici con l'età adulta, la sordità è progressiva, l’ipoacusia è simile a quella descritta per il tipo II ,ma compare attorno ai 3-5 anni e presenta una progressività nel corso degli anni (Kumar et al.. 1984).la perdita dell'udito e perdita della vista sono progressivi a partire dai primi decenni di vita a differenza delle altre forme di sindrome di Usher. Sebbene la maggior parte dei bambini con il disturbo hanno un equilibrio normale o quasi normale, alcuni possono sviluppare problemi di equilibrio in seguito, la disfunzione vestibolare si ha nella metà dei casi, i pazienti presentano un quadro variabile caratterizzato da progressivo peggioramento (Fishman. 1979: Fishman et al.. 1983). Con la mezza età, gli individui più colpiti sono sordi. Anche la perdita della vista causata dalla retinite pigmentosa si sviluppa nella tarda infanzia o nell'adolescenza. Le persone con sindrome di Usher di tipo III possono anche avere difficoltà con l'equilibrio a causa di problemi dell'orecchio interno. Questi problemi variano tra gli individui affetti, Nella s di Usher tipo III i bambini hanno un udito normale alla nascita, mentre i sintomi vestibolari e la retinite pigmentosa insorgono in età variabili
|
Quali geni sono legati alla sindrome di Usher?
Mutazioni nel CDH23, CLRN1, GPR98, MYO7A, PCDH15, USH1C, USH1G, ei geni USH2A causano la sindrome di Usher.
I geni legati alla sindrome di Usher forniscono istruzioni per fare proteine che svolgono ruoli importanti nella udito normale, equilibrio, e la visione. Funzionano nello sviluppo e nel mantenimento delle cellule ciliate, che sono cellule sensoriali dell'orecchio interno che aiutano a trasmettere segnali di movimento del suono e al cervello. Nella retina, questi geni sono coinvolti nel determinare la struttura e la funzione delle cellule sensibili alla luce bastoncelli e coni. In alcuni casi, l'esatto ruolo di questi geni in ascolto e visione è sconosciuta. La maggior parte delle mutazioni responsabili della sindrome Usher portare ad una perdita di cellule ciliate nell'orecchio interno e una graduale perdita di coni e bastoncelli della retina. La degenerazione di queste cellule sensoriali provoca perdita di udito, problemi di equilibrio, e la perdita di visione caratteristica di questa condizione.
Usher sindrome tipo che posso derivare da mutazioni nel CDH23, MYO7A, PCDH15, USH1C, o geneUSH1G. Almeno altri due geni identificati anche causare questa forma di sindrome di Usher.
La sindrome di Usher di tipo II è causata da mutazioni in almeno quattro geni. Solo due di questi geni,USH2A e GPR98 (chiamato anche VLGR1), sono stati identificati.
Le mutazioni in almeno due geni sono responsabili della sindrome di Usher tipo III; tuttavia, CLRN1 è l'unico gene che è stato identificato.
Per saperne di più sul CDH23 , CLRN1 , GPR98 , MYO7A , PCDH15 , USH1C , USH1G , e USH2Ageni.
Nella sindrome di Usher tipo I sono stati identificati 7 sedi di linkage e 4 geni (MYO7A. USH1C. CDH23. PCDH15) a carico dei quali sono state osservate varie mutazioni, con sordità congenita profonda, areflessia vestibolare bilaterale responsabile di un ritardo della deambulazione (deambulazione dopo 18 mesi) e retinite che si sviluppa durante l'infanzia. I primi segni visivi sono dei disturbi della visione in penombra, spesso verso l'inizio del secondo decennio di vita, ma l'esame sistematico del fondo dell'occhio può consentire la diagnosi molto prima di questa età, fin dai 3-4 anni. L'esame più precoce è l'elettroretinogramma, patologico prima dei primi segni nel fondo dell'occhio. La sindrome di Usher di tipo I è un'indicazione all'impianto cocleare precoce per ottenere una comprensione del linguaggio senza lettura labiale in questi bambini che, in età adulta, avranno una rilevante compromissione visiva. Porre la diagnosi attraverso l'esame del fondo dell'occhio a 4 anni è quindi già una situazione tardiva. In linea di principio l'esame oftalmologico esteso al fondo dell'occhio deve essere sistematico e ripetuto nel bambino e nell'adulto sordi, e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell'occhio è normale La funzionalità vestibolare è caratterizzata da areflessia nella sindrome di Usher Tipo I ,da normoreflessia nella sindrome di Usher Tipo II e da risposte che vanno progressivamente peggiorando nella sindrome di Usher Tipo III (Kumar et al. 1984: Möller et al.. 1989)
Nella sindrome di Usher tipo II sono state osservate varie mutazioni a carico dii gene (USH2A); tuttavia è verosimile che vi sia una eterogeneità genetica dal momento che sono state identificate 2 ulteriori sedi di linkage, la sordità è in media grave, non progressiva, predominante sulle frequenze acute, la retinite un poco più tardiva e i segni vestibolari assenti. Nella Usher tipo III è stato recentemente individuato 1 gene (USH3) a carico del quale sono state osservate varie mutazioni (Tab. III). la sordità è progressiva, i segni vestibolari e l'età di inizio della retinite sono variabili (per una rassegna, vedi [Grøndahl J (1987). 8]).
A tutt'oggi sono stati localizzati 12 geni diversi responsabili di queste forme Geni e le proteine che codificano sono stati identificati per 7 dei 12 loci., cinque per la sindrome di Usher tipo I, due per il tipo II e uno per il tipo III (Tabella II/III). MYO7A e CDH23 rappresentano rispettivamente il 30 e 29% dei casi di Usher tipo I e USH2A il 40% dei tipi II(Ouyang X.) La diagnosi molecolare non è eseguita di routine e la diagnosi è essenzialmente clinica.
I geni che causano la sindrome di Usher sono MY07A, USH1C, CDH23, PCDH15,e SANS, che causa USH1, USH2A (che causa USH2 ), e USH3A (che provoca USH3 ). Una mutazione, denominata R245X, del PCDH15 gene può rappresentare una grande percentuale di USH1 casi in Ashkenazi popolazione ebraica di oggi La maggior parte delle s. di Usher sono causate da mutazioni dei geni MYO7A, CDH23, USH2A che codificano per proteine importanti per la costituzione delle cellule cigliate e della matrice extracellulare della coclea.
|
|
|
|
|
|

Tab, II. Genetica della s, di Usher.
|
Locus |
|
Gene |
Marker |
Most lmportant Reference |
OMIM |
|
USH1A |
14q32 |
Unknown |
D14S250, D14S260, D14S292, D14S78 |
Kaplan et al., 1992 |
276900 |
|
USH1B |
11q135 |
MYO7A |
Di 1S906, Dl 1S911, D11S52, OMP-CA |
Weil et al,1995 |
276903 |
|
USH1C |
hp15.1 |
USH1C |
D11S902,D11S921, D11S899,D11S861 |
Smith et al,, 1992 |
276904 |
|
USH1D |
10q |
CDH23 |
D10S529, D10S202, D10S573 |
Wayne et al,, 1996 |
601067 |
|
USH1E |
21q |
Unknown |
D21S1884, |
Chaib et al, 1997 |
602097 |
|
USH1F |
10q2122 |
PCDH15 |
D10S199, D10S578, D10S596 |
Ahmed et aL, 2001 |
602083 |
|
USH1G |
17q24-25 |
Unknown |
|
Mustapha et al.., 2002 |
|
|
USH2A |
1q41 |
USH2A |
D1S229, D1S490, D1S237, D1S474 |
Kimberling et al, |
276901 |
|
USH2B |
3p2324..2, |
Unknown |
D3S1578, D3S3647, D3S3658 |
Hmani et al,, 1999 |
276905 |
|
USH2C |
5q14.3-q21.3 |
Unknown |
D5S428, D5S421 |
Pieke-Dahl et al., 2000 |
605472 |
|
USH 3 |
3q21-q25 |
USH3 |
D3S1299, D3S1555, D3S1280, D3S1 279 |
Sankila et al, 1995 Joensuu et al., 2001 |
276902 |
Geni associati con la sindrome di Usher Tab, III.
|
Tabella 1: geni collegati alla sindrome di Usher |
||||||||
|
Tipo |
Freq [ 16 ] |
Gene locus |
Gene |
Proteina |
Funzione |
Dimensioni (AA) |
UniProt |
|
|
USH1B |
39-55% |
11q 13.5 |
Myosin VIIA |
2215 |
||||
|
USH1C |
6-7% |
11p 15.1-p14 |
552 |
|||||
|
USH1D |
19-35% |
10q 21-q22 |
3354 |
|||||
|
USH1E |
raro |
21q 21 |
? |
? |
? |
? |
? |
|
|
USH1F |
11-19% |
10q 11.2-Q21 |
1955 |
|||||
|
USH1G |
7% |
17q 24-q25 |
SANS |
461 |
||||
|
USH2A |
80% |
1q 41 |
Usherin |
Transmembrane linkage |
5202 |
|||
|
USH2C |
15% |
5q 14.3-q21.1 |
VLGR1b |
Molto grande GPCR |
6307 |
|||
|
USH2D |
5% |
9q 32-q34 |
Whirlin |
907 |
||||
|
USH3A |
100% |
3q 21-q25 |
Clarin-1 |
Shaping Synaptic |
232 |
|||
Come viene diagnosticata la sindrome di Usher?
Poiché la sindrome di Usher colpisce l'udito, equilibrio, e la visione, la diagnosi del disturbo di solito include la valutazione di tutti e tre i sensi. Misure comportamentali e oggettive del sistema uditivo sono tecniche familiari per l'audiologo. Proprio come con le misure acustiche, la valutazione della funzione visiva può essere comportamentale (cioè, l'acuità e campo visivo) o oggettiva. Una videoelectronistagmografia (VNG) che misura i movimenti involontari degli occhi potrebbe evidenziare dei problemi di equilibrio. I tests obiettivi comprende l'esame diretto della retina, dove un oculista troverà vasi sanguigni scoloriti, un pallore cereo, e grumi di cellule retiniche morte chiamati spicole ossee (Carr & Noble, 1981). Tuttavia, questi risultati fisici non sono evidenti fino a ben dopo che il paziente è sintomatico. La prova definitiva di RP è un elettroretinogramma (ERG). Come una risposta uditivi del tronco encefalico, l'ERG è una risposta evocata ma dai coni e bastoncelli della retina. Poiché il test richiede l'inserimento di una lente a contatto matrice / elettrodo, anestesia generale è necessaria per i bambini, mentre farmaci topici possono essere utilizzati con gli adulti. Anche se un bambino può avere una visione ancora relativamente buona, l'ERG sarà ridotto o assente quando sindrome RP / Usher è presente. Vi è qualche evidenza che mentre la perdita di ampiezza del ERG è simile tra Usher tipo I e tipo II, il ritardo implicito si può distinguere tra i tipi (Seeliger, Zrenner, Apfelstedt-Sylla, e Jaissle, 2001). Un ERG presenta un vantaggio diagnostico distinto che sarà anormale molto prima dei segni fisici di morte cellulare (spicole ossee) che compaiono sulla retina. Questo spiega perché un oculista spesso non fa la diagnosi con un giovane paziente, in quanto il bambino si esibirà bene nel test dell'acuità visiva e le sue retine sotto esame diretto appariranno normali.
Valutazione degli occhi può includere un test del campo visivo per misurare la visione di una persona periferica, un elettroretinogramma (ERG) per misurare la risposta elettrica delle cellule fotosensibili dell'occhio, e un esame della retina per osservare la retina e altre strutture nella parte posteriore l'occhio. La diagnosi precoce della sindrome di Usher è molto importante. Prima che i genitori sanno che il loro bambino ha la sindrome di Usher, prima il bambino potrà iniziare programmi di formazione educativa speciali per gestire la perdita di udito e della vista.
Come le persone ereditano la sindrome di Usher? APPROFONDIMENTO
Questa condizione è ereditata come carattere autosomico recessivo, il che significa che entrambe le copie del gene in ogni cellula hanno mutazioni. I genitori di una persona con una malattia autosomica recessiva portano ciascuno una copia del gene mutato, ma in genere non mostrano segni e sintomi della malattia.
Dove posso trovare informazioni circa la diagnosi o la gestione della sindrome di Usher?
Tali risorse riguardano la diagnosi o il trattamento della sindrome di Usher e possono includere fornitori di trattamento.
- Gene Recensione: Sindrome di Usher di tipo 1

- Gene Recensione: Sindrome di Usher di tipo 1
- Gene Recensione: Sindrome di Usher di tipo 2
- Gene Recensione: Sindrome di Usher di tipo 2
- Genetica Registro Test: Retinite
pigmentosa Sindrome-sordità
- Genetica Registro Test: La sindrome di Usher,
tipo 1
- Genetica Registro Test: La sindrome di Usher,
tipo 1C
- Genetica Registro Test: sindrome di Usher,
tipo 1D
- Genetica Registro Test: La sindrome di Usher,
tipo 1E
- Genetica Registro Test: La sindrome di Usher,
tipo 1F
- Genetica Registro Test: La sindrome di Usher,
tipo 1G
- Genetica Registro Test: La sindrome di Usher,
tipo 2A
- Genetica Registro Test: La sindrome di Usher,
tipo 2C
- Genetica Registro Test: La sindrome di Usher,
tipo 3
- Genetica Registro Test: Usher sindrome tipo ID /
F, CDH23/PCDH15, digenica
- MedlinePlus Enciclopedia:
Retinite pigmentosa
È disponibile un test genetico per la sindrome di Usher?
Finora, sono stati trovati 11 loci genetici (un segmento del cromosoma in cui si trova un certo gene) per causare la sindrome Usher, e sono stati individuati nove geni che causano la malattia. Sono:
- Tipo 1 La sindrome di Usher: MY07A, USH1C, CDH23, PCDH15, SANS
- Tipo 2 La sindrome di Usher: USH2A, VLGR1, whrn
- Tipo 3 sindrome di Usher: USH3A
Con così tanti possibili geni coinvolti nella sindrome di Usher,i test genetici per la malattia non possono sono effettuati in modo capillare. La diagnosi di sindrome di Usher è di solito effettuata attraverso i tests dell'udito, dell'equilibrio, e della visione. Il test genetico per alcuni dei geni identificati è clinicamente disponibile. Per conoscere i laboratori che effettuano test clinici, visitare il sito Web www.GeneTests.org e cercare la directory laboratorio digitando il termine "sindrome di Usher". Il test genetico per ulteriori geni sindrome di Usher potrebbe essere disponibile attraverso studi di ricerca clinica. Per informazioni sulle sperimentazioni cliniche che includono il test genetico per la sindrome di Usher, visitare il sito Web www.clinicaltrials.gov e digitare il termine di ricerca "sindrome di Usher" o "test genetici Usher".
Condotta da tenere
L'esame oftalmologico esteso al fondo dell'occhio deve essere sistematico e ripetuto nel bambino e nell'adulto sordi e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell'occhio è normale. La diagnosi precoce è molto importante al fine del programma terapeutico-riabilitativo. La prognosi infatti è caratterizzata dalla comparsa di una cecità attorno ai 3 0-40 anni di età che andrà a sommarsi al deficit uditivo. compromettendo notevolmente il grado di autonomia del soggetto
Che trattamento è disponibile per la sindrome di Usher?
Attualmente non esiste un trattamento medico per prevenire, rallentare la progressione della, o inibire la trasmissione di sindrome di Usher. Impianto cocleare è una valida opzione provata per le persone con sindrome di Usher. Gli individui con tipo I sono candidati dalla nascita, mentre quelli con tipo II o III può diventare candidati nel corso del tempo. Qualità significativo di miglioramento della vita sono state dimostrate per i bambini con sordità congenita che ricevono impianti cocleari (Schorr, Roth, e Fox, 2009), e l'impianto è stato dimostrato come benefico per i bambini sordi con una varietà di disabilità associate (Berrettini et al., 2008). Perché la perdita dell'udito nella sindrome di Usher è cocleare in natura, l'impianto cocleare è una raccomandazione ragionevole e ha, di fatto, dimostrato di beneficiare funzionamento uditiva e sociale dei bambini con sindrome di Usher tipo I (Damon, Pennings, Snik, e Mylanus 2006 ; Liu et al, 2008)..
Ad oggi non esiste alcun trattamento per la RP. Molti individui mantengono un piccolo campo visivo utilizzabile anche in quinta o sesta decade di vita. Vi sono prove preliminari che i fattori ambientali e dietetici possono aumentare la longevità di visione utilizzabile. Questi includono proteggere la retina dalla luce ultravioletta indossando 100% UVA / UVB occhiali da sole e mantenere la salute globale ottimale. L'esercizio fisico e la dieta, come ad esempio a base di pesce ricco di omega-3, sono stati anche suggeriti (Berson, 2000). Gli studi sulla vitamina A e acido docosaesaenoico (DHA) trattamenti hanno creato qualche polemica (Massof & Finkelstein, 1993) e fino ad oggi sono stati solo condotti su persone con RP non sindromica e alcuni Usher di tipo II pazienti (Berson et al sentite., 1993, 2004a , 2004b). I pazienti devono essere avvertiti concernente una supplementazione di vitamina perché consumano troppo può causare ipervitaminosi conseguente cecità, mancanza di crescita, e anche la morte. Le donne e le donne incinte che possono diventare incinte non dovrebbero utilizzare integratori come la vitamina A, perché possono causare difetti alla nascita nel feto.
Implicazioni psicosociali della sindrome di Usher
La perdita sensoriale duale, in particolare la perdita progressiva della vista, presenta molteplici sfide per le persone con sindrome di Usher e le loro famiglie. In genere la perdita dell'udito è stata affrontata e le decisioni sono state fatte sulla metodologia di comunicazione. Una cultura identità Deaf può essere stata stabilita prima conoscenza della perdita della vista imminente. I genitori sono spesso sopraffatti dalla seconda diagnosi e credono che dovrebbero proteggere i loro figli da questa informazione fino a tardi. Nel frattempo, il bambino può sospettare qualcosa ed avere ancora più paura perché lui o lei non capisce. Condivisione di informazioni adeguate all'età con i bambini è il miglior approcio. Non hanno bisogno di sentirsi dire che stanno "diventando ciechi", ma hanno bisogno di sapere che vedono in modo diverso e che ci sono strategie che li aiuteranno a navigare nel loro mondo. Può essere giustificata l'aiuto di UNA consulenza professionale. Vivere con la sindrome di Usher richiede adattamenti per tutta la vita a cambiare lo stato di visione (Miner, 1995) e, in alcuni casi, cambiare le capacità uditive. Come audiologi, non possiamo supporre ci sono professionisti che hanno familiarità con l'impatto della perdita sensoriale duale. La maggior parte degli specialisti della vista non conoscono la perdita dell'udito e di fatto dipendono dall’audito per aiutare i loro pazienti non vedenti. Gli individui con sindrome di Usher sono sempre sordi o con problemi di udito prima che siano ipovedenti o non vedenti.
Il ruolo del audiologo
La diagnosi di una significativa perdita di udito in un bambino può così dominare la nostra attenzione come audiologi che possiamo trascurare le altre condizioni invalidanti. I genitori si affidano a audiologi per informazioni e supporto nel lavoro con i loro bambini che hanno la perdita dell'udito. Infatti, Steinberg e colleghi (2007) hanno identificato le interazioni dei genitori con l'audiologo come tema importante nel loro esame dei racconti dei genitori in materia di test genetici per la perdita dell'udito. Gli autori hanno indicato che le aspettative dei genitori con gli audiologi includono le informazioni riguardanti le risorse, sostegno e orientamento con i rinvii e test, e permettendo ai genitori di adattarsi alle loro nuovi ruoli. In sintesi, "audiologi sono spesso tenuti a consigliare, guidare e aiutare i genitori attraverso le loro decisioni e le scelte" (Steinberg et al., 2007, p. 64). Inoltre, l'American Speech-Language-Hearing Association (ASHA) Linee guida per la valutazione Audiologic dei bambini dalla nascita ai 5 anni di età consiglia di audiologi dovrebbero ", come opportuno, discutere valutazioni speciali aggiuntivi (ad esempio, la genetica, oculistica, sviluppo infantile) con i genitori / tutori ei neonati primario fornitore di cure "(ASHA, 2004, p. 19). Ciò richiede che l'audiologo avere familiarità con l'epidemiologia genetica della sordità e delle risorse di rinvio e di informazione.
Altre forme di ipoacusia associate a perdita della vista
Secondo la Gallaudet Research Institute (2008), il 40% di tutti i bambini con perdita dell'udito neurosensoriale sono noti per avere una o più condizioni invalidanti supplementari. In questo gruppo, il 10% ha problemi di vista non correggibile. Differenziazione tra i bambini ipovedenti e coloro che sono sordo-ciechi è irto di difficoltà dovute alla variabilità dell’udito residuo e della funzione visiva. Perdita della vista, come la perdita dell'udito, varia notevolmente in gravità e può essere misurata come acuità visiva da vicino e lontano, campo visivo periferico, sensibilità al contrasto, visione dei colori, sensibilità all'abbagliamento, e / o la visione notturna. Così come non si può veramente prevedere la competenza comunicativa di un paziente da un audiogramma per i toni- puri, un singolo parametro visivo non predice il funzionamento visivo.
Mentre Usher è la sindrome più frequentemente associato con RP, ci sono altre condizioni che il clinico deve conoscere. La Sindrome di Alport si manifesta con perdita dell’ udito, nefrite progressiva, e chiazze maculari che possono essere fraintesi come RP, mentre la malattia di Refsum si presenta con anosmia, sordità, e vero RP (Hamel, 2006). Altre condizioni come la sindrome di CHARGE, citomegalovirus, toxoplasmosi, e la meningite può causare contemporaneamente perdita dell’udito e della vista. 2008 Conte Child Nazionale dei bambini e dei giovani che sono sordo-ciechi riporta 10.766 bambini di età compresa nascita attraverso 21 anni sono stati identificati come (Consorzio Nazionale sulla sordocecità, 2008) sordo-ciechi. Indipendentemente dalla eziologia, un audiologo deve essere sensibile alla visione e le strategie suggerite qui per lavorare con i pazienti con sindrome di Usher è applicabile a molti altri pazienti con compromesse delle capacità visive.
Casi individuali da wikipendia
Casi individuali [ modifica ]
Una donna di 31 anni, con la sindrome di Usher, Rebecca Alexander, è stato profilato in Marie Claire nel novembre 2007. [21] Dopo la laurea presso la University of Michigan con ottimi voti, Alexander ha continuato alla Columbia University , dove ha conseguito due gradi di master nella sanità pubblica e clinico sociale. Rebecca è un membro attivo della sua comunità, collaborando con diverse associazioni di beneficenza a New York. La dedizione di Rebecca come membro attivo della sua comunità è stata in particolare riconosciuta quando è stata selezionata come una "Comunità Hero" per correre con la torcia olimpica per i Giochi di Atlanta olimpici del 1996 in onore del suo lavoro di volontariato per il progetto Open Hand, un'organizzazione no-profit consegna pasti per le persone che vivono con l'HIV / AIDS nella zona di San Francisco Bay. Rebecca ha ricevuto la sua formazione psicoterapia psicodinamica attraverso l'American Institute di Psicoanalisi. Attualmente lavora in uno studio privato specializzato nel trattamento dei disturbi dell'umore e d'ansia, disturbi alimentari, dipendenze, disabilità, e traumi. Mentre attualmente facilita seminari di gruppo per la Foundation Fighting Blindness durante le conferenze nazionali, Rebecca è anche in procinto di lanciare l'iniziativa Usher III, un'organizzazione no profit dedicata alla scienza e alla ricerca che cerca di trovare una cura per Usher III. Rebecca insegna ciclismo / spin nelle classi interne con un forte seguito in alcune palestre di New York City. È stata descritta sulla Today Show della NBC il 20 marzo 2009, che è stato nominato per un premio Emmy nel settembre 2010. Lei è la sorella di NBC News Nazionale Corrispondente Peter Alexander.
Christine "Coco" Roschaert [22] è una persona conosciuta con la sindrome di Usher. Ha pubblicato un video blog su YouTube, [23] e recentemente è stato l'altoparlante kick-off per la Settimana della prevenzione della sordità presso l' Università del Vermont . [24]Nel 2006, si è laureata con una laurea in Scienze della Comunicazione alla Gallaudet University ; lì, lei ha fatto lo sciopero della fame come segno di protesta 2006 organizzata dalla Gallaudet Stati Ora Movimento . [25] Roschaert è ora in Nigeria per la fondazione del programma prima sordociechi .
Una web-community, UsherLife , delle persone con sindrome di Usher è stata fondata il 1 ° febbraio 2005 da Nick Sturley. Anche se centrata sulla Gran Bretagna , che offre risorse per tutte le persone con sindrome di Usher. L'organizzazione sta ospitando regolarmente ritrovi in Inghilterra, come il Usher Hood Pub a Nottingham [26] e un viaggio a Brighton Pier. [27] Altre persone con sindrome di Usher hanno inviato video sulla loro vita e le condizioni su YouTube , in particolare . Ginny Paja-Nyholm[28] Nell'ottobre 2007, Candice, una mamma che vive in Texas, ha iniziato a bloggare sulle sue due figlie, Jasmine e Rebecca;Rebecca che hanno la sindrome di Usher I. [29]
Catherine Fischer ha scritto un'autobiografia ben accolta di “crescere con la sindrome di Usher in Louisiana”, intitolato Orchidea del Bayou. [30] Allo stesso modo, Vendon Wright ha scritto due libri che descrivono la sua vita con la sindrome di Usher, ero cieco, ma ora vedo [31] e attraverso i miei occhi. [32] Louise Boardman ha anche scritto un breve libro intitolato Mio figlio ha la sindrome di Usher. [33]
Christian Markovic, un artista che vive con la sindrome di Usher, gestisce una società, Fuzzy disegni wuzzy. [34]
Spencer Tracy figlio s 'Giovanni era una persona ben conosciuta con la sindrome di Usher, che ha vissuto una vita piena. [35] IlJohn Tracy Clinic è stata fondata nel 1942 da sua madre Louise per offrire assistenza gratuita ai genitori dei bambini udenti e bambini in età prescolare . [1]
Jacob Desormeaux, figlio del fantino di ippica Kent Desormeaux , ha la sindrome di Usher. Jacob è nato sordo e sta progressivamente diventando cieco. Kent ha dedicato la sua corsa nelle Belmont Stakes , che lui e il suo cavallo darebbero Big Brown la Triple Crown, a suo figlio Giacobbe. La famiglia ha avviato un'organizzazione per raccogliere fondi e la consapevolezza della malattia. La sindrome di Usher è sproporzionatamente comune tra i cajun della Louisiana a sud, come Desormeaux e Fischer, a causa di una mutazione genetica tra i primi francesi Acadian coloni in Nova Scotia .
Elica del DNA co-scopritore e premio Nobel James D. Watson ha pubblicato[36] USH1B mutazioni omozigoti, secondo il suo genoma. Non è chiaro il motivo per cui egli non ha sviluppato la sindrome. Questa mancanza di penetranza genetica sostiene che l'espressione del fenotipo della sindrome di Usher può essere più complessa di quanto inizialmente ipotizzato.
Il "Nalaga'at" israeliano (fare touch) sordo-ciechi Acting Ensemble è composto da 11 attori sordo-cieco, molti dei quali sono diagnosticati con sindrome di Usher. Il gruppo teatrale ha messo su diverse produzioni ed è apparso sia localmente che in Israele e all'estero a Londra e Broadway. [37]
References
Alexander R, Grossman AJ (November 2007). "out of sight, out of sound". Marie Claire 14 (11): 191–193.American Speech-Language-Hearing Association. (2004). Guidelines for the audiologic assessment of children from birth to 5 years of age. Available from www.asha.org/policy .
Bell J (1933). Retinitis Pigmentosa and Allied Diseases (2nd ed.). London: Cambridge University Press.
Berrettini, S., Forli, F., Genovese, E., Santarello, R., Arslan, E., Chilosi, A., & Cipriani, P. (2008). Cochlear implantation in deaf children with associated disabilities: Challenges and outcomes. International Journal of Audiology, 47, 199–208.
Berson, E. (2000). Nutrition and retinal degeneration. International Ophthalmology Clinics , 40 (4), 93–111.
Berson, E., Rosner, B., Sandberg, M., Hayes, K., Nicholson, B., Weigel-DiFranco, C., & Willett, W. (1993). A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Archives of Ophthalmology, 111 , 761–772.
Berson, E., Rosner, B., Sandberg, M., Weigel-DiFranco, C., Moser, A., Brockhurst, R., et al. (2004a). Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Archives of Ophthalmology, 122 , 1297–1305.
Berson, E., Rosner, B., Sandberg, M., Weigel-DiFranco, C., Moser, A., Brockhurst, R., et al. (2004b). Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Archives of Ophthalmology, 122, 1306–1314.
Boughman, J., Vernon, M., & Shaver, K. (1983). Usher syndrome: Definition and estimate of prevalence from two high-risk populations. Journal of Chronic Diseases, 36, 595–603.
Carr, R., & Noble, K. (1981). Retinitis pigmentosa. Ophthalmology, 88 (2), 169–172.
Chen, D. (2004). Young children who are deaf-blind: Implications for professional in deaf and hard of hearing services. Volta Review, 104 (4), 273–284.
Cohen, M., Bitner-Glindziez, M., & Luxon, L. (2007). The changing face of Usher syndrome: Clinical implications. International Journal of Audiology, 46, 82–93.
Damon, G., Pennings, R., Snik, A., & Mylanus, E. (2006). Quality of life and cochlear implantation in Usher syndrome type I. Laryngoscope, 116, 723–728.
Davenport S, Omenn G (1977). The Heterogeneity of Usher Syndrome (volume 426 ed.). Amsterdam: Excerpta Medica Foundation.
Edwards, A., Fishman, G., Anderson, R., Grove, S.,
& Derlackie, D. (1998). Visual acuity and visual field impairment in
Usher syndrome. Archives of Ophthalmology, 116 , 165–168.
Fishman GA, Kumar A, Joseph ME, Torok N, and Andersonj RJ (1983). "Usher's syndrome". Archives of Ophthalmology 101 (9): 1367–1374.
Gallaudet Research Institute. (2008). Regional and national summary report of data from the 2007–08 annual survey of deaf and hard of hearing children and youth. Washington, DC: Author.
Gerber, S; Bonneau, D; Gilbert, B; Munnich, A; Dufier, JL; Rozet, JM; Kaplan, J (2006). "USH1A: chronicle of a slow death". American Journal of Human Genetics 78 (2): 357–9.
Gorlin R, Tilsner T, Feinstein S, Duvall AJ (1979). "Usher syndrome type III". Arch. Otolaryngol. 105 (6): 353–354.
Grøndahl J (1987). "Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway". Clin. Genet. 31 (4): 255–264.
Gorlin, R., Torella, H., & Cohen, M. (1995). Hereditary hearing loss and its syndromes. Oxford, England: Oxford University Press
Hallgren B (1959). "Retinitis pigmentosa combined with congenital deafness with vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases: Clinical and geneto-statistical survey". Acta Psychiatrica Scandinavica Suppl. 34 (138): 9–101.
Hamel, C. (2006). Retinitis pigmentosa. Orphanet Journal of Rare Diseases, 1 :(40, 1 – 12).
Hammerschlag V (1907). "Zur Kenntnis der hereditaer-degenerativen Taubstummen und ihre differential diagnostische Bedeutung". Z. Ohrenheilk. 54: 18–36.
Harrison, S., & Rousch, J. (1996). Age of suspicion, identification, and intervention for infants and young children with hearing loss: A national study. Ear and Hearing, 17, 55–62.
Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM, Yang XJ, Williams DS (2007). "Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B". Gene Therapy 14 (7): 584–594
Hicks, W., & Hicks, D. (1981). The Usher's syndrome adolescent: Programming implications for school administrators, teachers, and residential advisors. American Annals of the Deaf, 26 , 422–431.
Hope CI, Bundey S, Proops D, Fielder AR (1997). "Usher syndrome in the city of Birmingham — prevalence and clinical classification". British Journal of Ophthalmology 81 (1): 46–53.
Innaccone, A., Kritchevsky, S., Ciccarelli, M., Tedesco, S., Macahuso, C., Kimberling, W., & Somes, G. (2004). Kinetics of visual field loss in Usher syndrome type II. Investigative Ophthalmology & Visual Science, 45 , 784–792.
Keats, B. (2002). Genes and syndromic hearing loss. Journal of Communication Disorders, 33, 355–366.
Kimberling, W., & Lindenmuth, A. (2007). Genetics, hereditary hearing loss, and ethics. Seminars in Hearing, 28 (3), 216–225.
Liebreich R (1861). "Abkunft aus Ehen unter Blutsverwandten als Grund von Retinitis pigmentosa". Dtsch. Klin. 13: 53.
Liu, X., Angeli, S., Rajput, K., Yan, D., Hodges, A., Eshraghi, A., et al. (2008). Cochlear implantation in individuals with Usher type 1 syndrome. International Journal of Pediatric Otorhinolaryngology, 72, 841–847.
Massof, R., & Finkelstein, D. (1993). Supplemental vitamin A retards loss of ERG amplitude in retinitis pigmentosa. Archives of Ophthalmology, 111, 751–754.
Merin S, Auerbach E (1976). "Retinitis pigmentosa". Surv. Ophthalmol. 20 (5): 303–345. doi:10.1016/S0039-6257(96)90001-6
Mets MB, Young NM, Pass A, Lasky JB (2000)."Early diagnosis of Usher syndrome in children".Transactions of the American Ophthalmological Society 98: 237–45.
Miner, I. (1995). Psychosocial implications of Usher syndrome, type I, throughout the life cycle. Journal of Visual Impairment & Blindness, 89 (3), 287–296.
Moller, C., Kimberling, W., Davenport, S., Priluck, L., & White, V. (1989). Usher syndrome: An otoneurologic study. Laryngoscope, 99, 73–79.
National Consortium on Deaf-Blindness. (2008). National Child Count of Children and Youth Who Are Deaf-Blind . Retrieved December 2, 2009, from www.nationaldb.org/documents/products/2008-Census-Tables.pdf [PDF].
Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C (2001). "A new clinical classication for Usher's syndrome based on a new subtype of Usher's syndrome type I". Laryngoscope 111 (1): 84–86.
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton V, Brown SD, Balkany T, Liu XZ (2005). "Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population". Hum Genet 116 (4): 292–299.
Pakarinen L, Tuppurainen K, Laipapala P, Mäntyjärvi M, Puhakka H (1996). "The ophthalmological course of Usher syndrome type III". International Ophthalmology 19 (5): 307–311.
Petit, C (2001). "Usher syndrome: from genetics to pathogenesis". Annual review of genomics and human genetics 2: 271–97.
Piazza, L., Fishman, G., Farer, M., Derlacki, D., & Anderson, R. (1986). Visual acuity loss in patients with Usher's syndrome. Archives of Ophthalmology, 104 , 1336–1339.
Plantinga, R., Pennings, R., Huygen, P., Sankila, E., Tuppurainen, K., Kleemola, L., et al. (2006). Visual impairment in Finnish Usher syndrome type III. Acta Ophthalmologica Scandinavica, 84 (1), 36–41.
Reiners, J; Nagel-Wolfrum, K; Jürgens, K; Märker, T; Wolfrum, U (2006). "Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease". Experimental eye research 83 (1): 97–119.
Reisser, C., Kimberling, W., & Otterstedde, C. (2002). Hearing loss in Usher syndrome type II is nonprogressive. Annals of Otology, Rhinology and Laryngology,111, 1108–1111.
Roux AF, Faugere V, Le Guedard S, Pallares-Ruiz N, Vielle A, Chambert S, Marlin S, Hamel C, Gilbert B, Malcolm S, Claustres M (2006). "Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%". J Med Genet 43 (9): 763–768.
Sadeghi, A., Eriksson, K., Kimberling, W., Sjostrom, A., & Moller, C. (2006). Longterm visual prognosis in Usher syndrome types 1 and 2. Acta Ophthalmologica Scandinavica, 84, 537–544.
Sadeghi, M., Cohn, E., Kelly, W., Kimberling, W., Tranebjarg, L., & Moller, C. (2004). Audiological findings in Usher syndrome types IIa and II (non-IIa). International Journal of Audiology, 43, 136–143.
Sadeghi, M., Cohn, E., Kimberling, W., Tranebjarg, L., & Moller, C. (2005). Audiological and vestibular features in affected subject with USH3: A genotype/phenotype correlation. International Journal of Audiology, 44, 307–316.
Sankila EM, Pakarinen H, Kääriäinen H, Aittomäki K, Karjalainen S, Sistonen P, de la Chapelle A (1995). "Assignment of Usher syndrome type III (USH3) gene to chromosome 3q". Hum. Mol. Genetics 4 (1): 93–98.
Schorr, E., Roth, F., & Fox, N. (2009). Quality of life for children with cochlear implants: Perceived benefits and problems and the perception of single words and emotional sounds . Journal of Speech, Language, and Hearing Research, 52, 141–152.
Seeliger, M., Zrenner, E., Apfelstedt-Sylla, E., & Jaissle, G. (2001). Identification of Usher syndrome subtypes by ERG implicit time. Investigative Ophthalmology and Visual Science, 42, 3066–3071.
Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, et al. (1994). "Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium".American Journal of Medical Genetics 50 (1): 32–38.
Steinberg, A., Kaimal, G., Ewing, R., Soslow, L., Lewis, K., Krantz, I., & Li, Y. (2007). Parental narratives of genetic testing for hearing loss: Audiologic implications for clinical work with children and families. American Journal of Audiology, 16, 57–67.
Usher C (1914). "On the inheritance of Retinitis pigmentosa with notes of cases". Roy. Lond. Ophthalmol. Hosp. Rep. 19: 130–236.Wagenaar, M., Snik, A., Kimberling, W., & Cremers, C. (1996). Carriers of Usher syndrome type IB: Is audiometric identification possible? The American Journal of Otology, 17, 853–858.
Vernon M (1969). "Usher's syndrome — deafness and progressive blindness. Clinical cases, prevention, theory and literature survey". Journal of Chronic Diseases 22 (3): 133–151.
von Gräfe A (1858). "Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der Netzhaut". Archiv für Ophthalmologie 4: 250–253.
Wallber, J. (1995–2005). Annual reports of the Eye & Ear Clinic. Rochester, NY: National Technical Institute for the Deaf at Rochester Institute of Technology.
Williams DS (2007). "Usher syndrome: Animal models, retinal function of Usher proteins, and prospects for gene therapy". Vision Research xx (3): xx–xx.doi:10.1016/j.visres.2007.08.015Web Resources
Boys Town National Research Hospital Usher Syndrome Web page
National Consortium on Deaf-Blindness
National Consortium on Deaf-Blindness, State Deaf-Blind Projects
National Institute on Deafness and Other Communication Disorders Usher Syndrome Web page
National Institutes of Health Usher Syndrome Web page
National Library of Medicine Genetics Home Reference
Second Sight Medical Products, Inc. retinal prosthesis—investigational device
Understanding Usher Syndrome: Information for School Counselors [PDF, 2 MB]






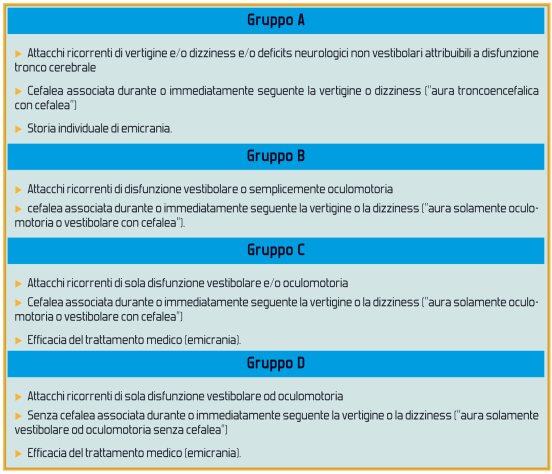









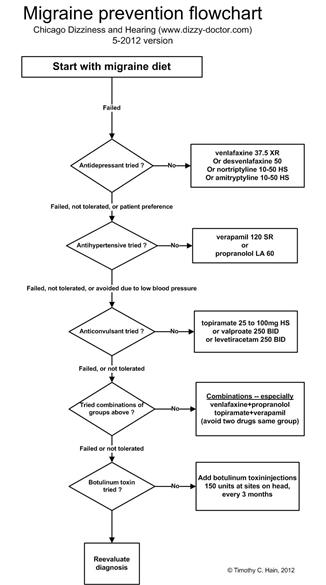
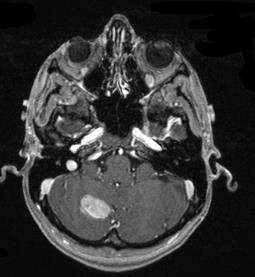
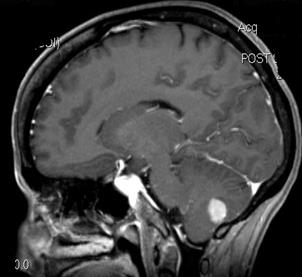

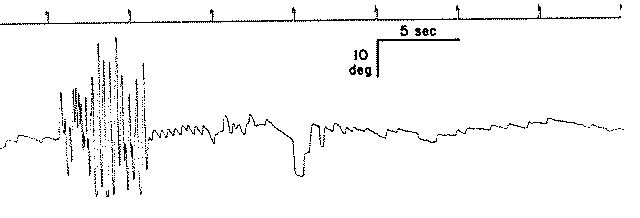




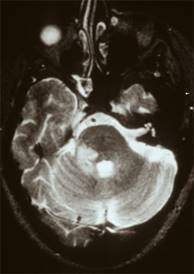
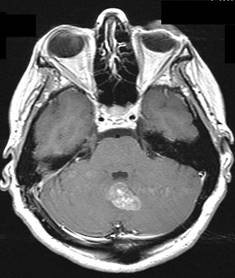


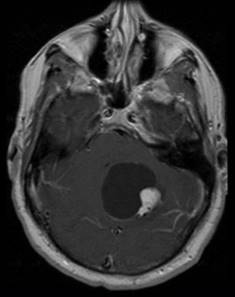

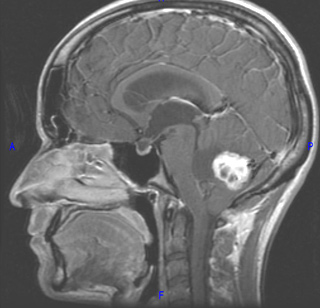

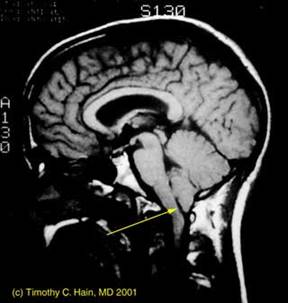
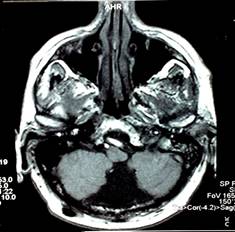

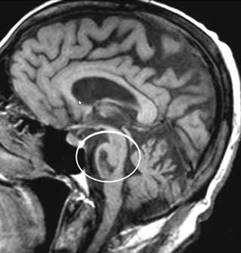
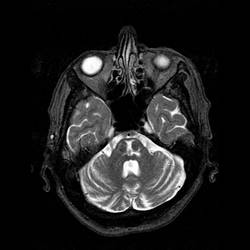


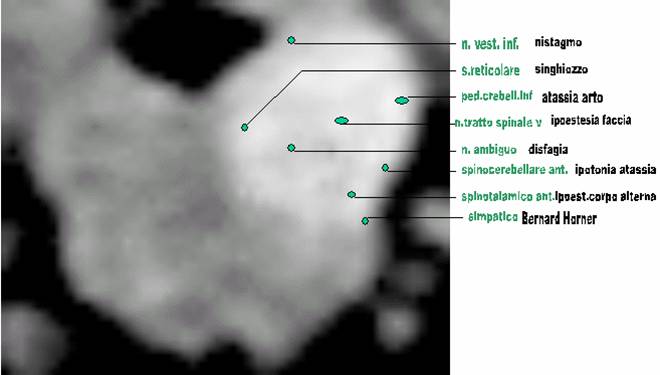



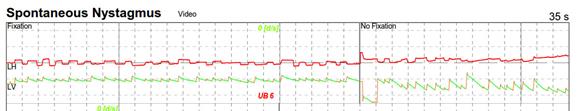


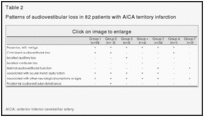









































{kind=link}